Orthogroup Conservation Analysis in Plants: Methods, Applications, and Biomedical Implications
Orthogroup conservation analysis has emerged as a powerful computational framework for identifying evolutionarily conserved gene families across plant species, overcoming challenges posed by frequent gene duplications and whole-genome duplication events.
Orthogroup Conservation Analysis in Plants: Methods, Applications, and Biomedical Implications
Abstract
Orthogroup conservation analysis has emerged as a powerful computational framework for identifying evolutionarily conserved gene families across plant species, overcoming challenges posed by frequent gene duplications and whole-genome duplication events. This article provides a comprehensive resource for researchers and scientists, exploring the foundational concepts of orthology, detailing cutting-edge methodologies like Orthologous Marker Gene Groups (OMGs) and OrthoFinder, and addressing common troubleshooting scenarios. We validate these approaches through comparative case studies across diverse plant families including Asteraceae and Oleaceae, demonstrating how orthogroup analysis reveals conserved cellular identities and adaptive evolutionary mechanisms. The insights gained have significant implications for understanding plant resilience mechanisms that could inform biomedical and clinical research strategies.
Understanding Orthogroups: Evolutionary Concepts and Genomic Significance in Plants
The concepts of orthology and paralogy, introduced by Walter Fitch in 1970, provide an essential evolutionary framework for comparative genomics [1]. While orthologs are homologous genes originating from speciation events and paralogs are those arising from gene duplication events, their functional implications are more nuanced than initially assumed [2] [3]. This guide objectively compares the performance of modern orthology inference methods and their applications in plant genomic research, focusing on orthogroup conservation analysis. We evaluate experimental protocols, computational tools, and database resources that enable researchers to tackle the complexities introduced by polyploidization, alternative splicing, and functional divergence in plant systems.
Historical Foundation and Conceptual Evolution
Original Definitions and Their Lasting Impact
Walter Fitch first distinguished orthologs from paralogs in his seminal 1970 paper, creating a conceptual cornerstone for modern genomics [1]. He proposed that orthologous genes (ortho = exact) are those where the homology results from speciation so that the history of the gene reflects the history of the species, while paralogous genes (para = in parallel) are those where the homology results from gene duplication so that both copies have descended side by side during the history of an organism [1] [2]. Fitch crisply articulated that "phylogenies require orthologous, not paralogous, genes," establishing a fundamental principle for evolutionary biology [1].
The diagram below illustrates the key evolutionary events that give rise to orthologous and paralogous relationships between genes:
Modern Conceptual Challenges and Complexities
The straightforward classification of homologs has evolved to accommodate complex evolutionary scenarios encountered in genomic research. Co-orthologs describe situations where a gene has multiple orthologs in another genome due to lineage-specific duplications, creating one-to-many or many-to-many relationships [3]. The additional distinction between in-paralogs (paralogs that duplicated after a given speciation event) and out-paralogs (paralogs that duplicated before a given speciation event) further refines these relationships for practical genomic analysis [3].
The original genocentric definition has also been challenged by discoveries in molecular biology. Between-species differences in protein domain architectures, the pervasiveness of alternative splicing in multicellular eukaryotes, and the presence of repetitive promiscuous domains have complicated orthology assignments, suggesting that evolutionary stable units may sometimes be smaller than entire genes [3].
Methodological Approaches for Orthology Inference
Computational Frameworks and Algorithms
Orthology inference methods predominantly fall into two categories: graph-based (pairwise alignment) methods and tree-based (phylogenetic) methods [4]. Each approach offers distinct advantages and limitations for different research contexts as summarized in the table below:
Table 1: Comparison of Major Orthology Inference Methods
| Method Type | Key Principle | Representative Tools/Databases | Accuracy Considerations | Computational Demand | Ideal Use Cases |
|---|---|---|---|---|---|
| Graph-based | Reciprocal Best Hits (RBH) | InParanoid, OrthoMCL, EnsemblCompara | Sensitive to incomplete genomes; may miss distant homologs | Lower resource requirements | Initial genome annotation; large-scale comparisons |
| Tree-based | Gene tree/species tree reconciliation | OrthoFinder, PhylomeDB, OrthologID | Higher accuracy but dependent on alignment and tree quality | Computationally intensive; scales with gene family size | Detailed evolutionary studies; complex gene families |
| Hybrid | Combines sequence similarity with additional constraints | PLAZA, OrthoDB, PlantOrDB | Balanced approach leveraging multiple evidence types | Moderate to high depending on implementation | Comprehensive genomic databases; cross-species comparisons |
The Orthologous Marker Groups (OMG) Method for Plant Single-Cell Transcriptomics
Recent innovations have addressed the specific challenges of orthology inference in plant species, which are complicated by frequent whole-genome duplications and tandem gene duplications. The Orthologous Marker Gene Groups (OMG) method enables cell type identification across diverse plant species without requiring cross-species data integration [5].
The OMG workflow involves three key stages:
- Marker Identification: Top N marker genes (N=200) are identified for each cell cluster in each species using standard tools like Seurat [5]
- Orthogroup Construction: OrthoFinder is employed to generate orthologous gene groups across multiple plant species (typically 15 species) [5]
- Statistical Testing: Pairwise comparisons using overlapping OMGs between clusters with Fisher's exact test to determine clusters with significant numbers of shared OMGs (FDR < 0.01) [5]
This method has been validated successfully in comparative analyses of tomato and Arabidopsis roots, where it identified 24 pairs of clusters with significant numbers of shared OMGs, accurately matching published cell type annotations [5]. The method demonstrates particular strength in handling one-to-many and many-to-many orthologous relationships common in plants.
Experimental Data and Performance Comparison
Testing the Ortholog Conjecture: Functional Conservation Evidence
The fundamental assumption that orthologs consistently retain greater functional similarity than paralogs (the "ortholog conjecture") has been systematically tested using comparative functional genomic data. A landmark study examining experimentally derived functions of more than 8,900 human and mouse genes revealed surprising patterns [6]:
Table 2: Functional Similarity Between Orthologs and Paralogs
| Comparison Type | Sequence Identity Range | Functional Similarity (Biological Process) | Functional Similarity (Molecular Function) | Key Findings |
|---|---|---|---|---|
| Orthologs | 51%-99% | 0.4-0.5 (no correlation with sequence identity) | 0.6-0.7 (no correlation with sequence identity) | Functional similarity remains constant regardless of sequence divergence |
| Paralogs | 51%-99% | Steep decline with decreasing identity | Moderate decline with decreasing identity | Positive correlation between sequence identity and functional similarity |
| Same-species paralogs | Various | Higher than orthologs at equivalent sequence identities | Higher than orthologs at equivalent sequence identities | Paralogs often better predictors of function than orthologs |
These findings challenge the straightforward functional transfer between orthologs, suggesting that cellular context plays a crucial role in functional evolution [6]. The study also found that paralogous pairs residing on the same chromosome are more functionally similar than those on different chromosomes, potentially due to higher levels of interlocus gene conversion [6].
Orthology Database Performance and Coverage
Multiple databases provide precomputed orthology relationships, each with different strengths in taxonomic coverage and methodology:
Table 3: Comparison of Orthology Database Resources
| Database | Methodology | Plant Species Coverage | Key Features | Limitations |
|---|---|---|---|---|
| PlantOrDB | Phylogenetic | 35 land plants + 6 green algae | Interactive visualization; diagnostic characters; query sequence placement | Limited to available sequenced genomes |
| OrthoDB | Graph-based | Multiple kingdoms | Evolutionary hierarchy of orthologs | Less specialized for plant-specific duplications |
| PLAZA 3.0 | Graph-based | 31 plant species | Integrated omics data; functional annotations | Cannot display very large gene families |
| EnsemblPlants | Graph-based | 38 plant species | Genome browser integration | Basic orthology inference methods |
| InParanoid | Graph-based | 100 species across kingdoms | Focus on in-paralogs | Limited browsing capabilities |
| PhylomeDB | Phylogenetic | 1,059 species across kingdoms | Comprehensive phylogenetic trees | Computational intensive to update |
PlantOrDB exemplifies specialized resources for plant research, containing 1,291,670 peptide sequences clustered into 49,355 homologous gene families with phylogenetic trees and speciation/duplication events identified for each node [4].
Applications in Plant Genomic Research
Orthogroup Conservation Analysis Across Plant Species
Orthology analysis has proven particularly valuable in plant genomics due to the prevalence of whole-genome duplication events in plant evolutionary history. The OMG method has enabled comparison of cell types across 15 diverse plant species, revealing 14 dominant groups with substantial conservation in shared cell-type markers across monocots and dicots [5]. This approach successfully mapped cell clusters from approximately 1 million cells across 268 cell clusters, demonstrating scalability to large datasets [5].
Conservation analysis also extends to regulatory mechanisms. Studies of alternative splicing in five Panax species revealed conserved molecular features of alternative splicing events despite significant expansion of AS events in Panax species compared to the outgroup Daucus carota [7]. Both skipped exons and retained introns showed conserved patterns of GC content and length biases across species of different ploidy levels, suggesting these sequence traits were established in the ancestral Panax lineage [7].
Cross-Species Functional Prediction and Drug Discovery
The transfer of functional information using orthology relationships has important applications in pharmacological research. The field of pharmacophylogenomics integrates molecular phylogeny with phytochemical profiles and bioactivities to identify medicinal compounds [8]. This approach operates on the principle that healing plants from related taxonomic groups are more likely to possess analogous chemical profiles and efficacies, a concept validated through studies of Scutellaria, Polygonateae, and Arnebia species [8].
Interestingly, studies of human-disease-associated gene orthologs in plants have revealed that 79.4% of 34 identified homologs in Arabidopsis are senescence-associated genes, suggesting a close relationship between human diseases and cellular senescence [9]. Protein-protein interaction network analysis showed that these genes formed two main subnetworks, interacting with multiple senescence-associated genes, indicating that leaf senescence may offer a model system for studying human disease pathogenesis [9].
Experimental Protocols for Orthology Analysis
Standard Workflow for Orthogroup Identification and Analysis
A typical experimental pipeline for orthology analysis in plant species involves sequential steps of data collection, processing, and interpretation as illustrated below:
Detailed Methodological Considerations
For orthology analysis in plant species, specific parameters and considerations include:
Sequence Datasets: Use high-quality annotated genomes from Phytozome, EnsemblPlants, or specialized databases. For Panax species studies, researchers used public transcriptome data from leaf and root tissues for five representative species and the outgroup Daucus carota [7].
Orthogroup Delineation: Apply OrthoFinder with default parameters for most applications, though adjusting inflation parameters for Markov clustering may be necessary for specific gene families. The OMG method utilized OrthoFinder with 15 plant species to generate orthologous groups [5].
Statistical Validation: Implement Fisher's exact test with FDR correction for OMG overlap analysis. In the OMG method, N=200 marker genes per cluster provided optimal balance between sensitivity and specificity [5].
Functional Annotation: Integrate Gene Ontology terms, KEGG pathways, and expression data from public repositories like NCBI's SRA database for cross-validation [9].
Research Reagent Solutions
Table 4: Essential Research Reagents and Resources for Orthology Studies
| Resource Category | Specific Tools/Databases | Primary Function | Application Context |
|---|---|---|---|
| Genomic Databases | Phytozome, EnsemblPlants, NCBI Genome | Source of annotated genome sequences and annotations | Data retrieval for initial analysis |
| Orthology Databases | PlantOrDB, PLAZA, OrthoDB, InParanoid | Precomputed orthology relationships | Validation and comparison of results |
| Analysis Tools | OrthoFinder, FastTree, DIAMOND, BLAST | Computational inference of orthologous relationships | Core analysis pipeline implementation |
| Specialized Plant Resources | Arabidopsis Information Resource (TAIR), Rice Annotation Project | Species-specific functional annotations | Functional transfer and validation |
| Visualization Platforms | OMG Browser, PhyD3, iTOL | Interactive visualization of orthology relationships | Data interpretation and presentation |
The concepts of orthology and paralogy introduced by Walter Fitch remain foundational to comparative genomics nearly five decades after their proposal. However, modern genomic research has revealed substantial complexity in the relationship between evolutionary history and gene function. While orthology analysis provides an essential framework for comparative studies, particularly in plant genomics where whole-genome duplications are common, the assumption that orthologs consistently maintain equivalent functions requires careful validation. The development of methods like OMG for plant single-cell transcriptomics and specialized databases like PlantOrDB demonstrate how evolutionary concepts can be adapted to address specific biological questions. As genomic data continue to accumulate, orthology analysis will remain indispensable for tracing evolutionary relationships, but researchers must incorporate multiple lines of evidence—including expression patterns, protein interactions, and functional assays—to accurately infer gene function across species.
In comparative plant genomics, accurately identifying homologous genes is fundamental to understanding evolutionary relationships, gene function, and phenotypic diversity. Two primary concepts form the cornerstone of this analysis: orthogroups and one-to-one orthologs. An orthogroup represents the set of all genes descended from a single gene in the last common ancestor of all species being considered, thereby encompassing both orthologs and paralogs [10]. In contrast, one-to-one orthologs refer to specific pairs of genes between two species where each gene has only a single direct descendant in the other species, indicating no lineage-specific duplications have occurred since their divergence [11]. This distinction is particularly critical in plant genomics due to the high frequency of whole-genome duplication (WGD) events and polyploidization, which create complex gene families that challenge simplified orthology inference methods [12]. The inherent biases in sequence-based methods, such as gene length dependency that adversely affects clustering accuracy, further complicate the landscape [10]. This guide objectively compares the performance of different orthology inference approaches when confronting these plant-specific challenges, providing researchers with a framework for selecting appropriate methodologies based on empirical evidence.
Key Concepts and Computational Methodologies
Orthogroup Inference: Capturing Complete Gene Families
Orthogroup inference methods aim to reconstruct complete gene families by clustering genes from multiple species based on sequence similarity. The OrthoFinder algorithm exemplifies this approach, employing a sophisticated workflow that begins with all-versus-all sequence similarity searches using tools like DIAMOND or BLAST [13] [10]. To address the significant gene length bias inherent in raw BLAST scores—where short sequences cannot achieve high scores while long sequences generate many high-scoring hits—OrthoFinder implements a novel score normalization procedure. This method transforms bit scores based on sequence length and phylogenetic distance, effectively eliminating the length dependency that plagues other methods and dramatically improving clustering accuracy [10]. Following score normalization, OrthoFinder applies the MCL clustering algorithm to identify orthogroups as highly connected regions within the sequence similarity network [10]. A major advancement in orthogroup inference is the implementation of phylogenetic hierarchical orthogroups, which are identified by analyzing rooted gene trees and provide orthogroup definitions at each hierarchical level (node) in the species tree. According to Orthobench benchmarks, these phylogenetically-informed orthogroups are 12-20% more accurate than graph-based clustering methods used previously [14].
One-to-One Ortholog Inference: Stringent Pairwise Relationships
One-to-one ortholog inference focuses on identifying specific pairwise relationships between genes in two species. The OMA (Orthologous Matrix) algorithm represents a comprehensive approach to identifying these relationships, beginning with the inference of pairwise orthologs based on sequence similarity between genomes [11]. Unlike orthogroups, pairwise orthologs specifically document the relationship cardinality between genes, with one-to-one (1:1) orthology indicating that both genes in the pair have only one ortholog in the other species [11]. The OMA method further identifies OMA Groups, defined as cliques of orthologs in the orthology graph where all genes are connected to each other by pairwise orthologous relations. While sometimes misunderstood as groups of 1:1 orthologs, OMA Groups may actually contain only one representative from co-orthologous groups, maintaining the property that all members are orthologous to all other members [11]. Additionally, OMA infers Hierarchical Orthologous Groups (HOGs), which are sets of genes descended from a common ancestral gene at a specific taxonomic level. These HOGs exhibit a nested structure, with groups defined at recent clades encompassed within larger groups defined at older clades [11].
Comparative Workflow: From Sequences to Orthology Assignments
The fundamental difference between these approaches lies in their starting points and objectives. Orthogroup methods begin with multiple species simultaneously and aim to reconstruct complete gene families, while one-to-one ortholog methods typically start with pairwise comparisons before potentially extending to multiple species. The following workflow diagram illustrates the key steps in each approach:
Plant-Specific Challenges in Orthology Inference
Prevalence of Polyploidy and Whole-Genome Duplications
Plant genomes present unique challenges for orthology inference due to their exceptional evolutionary dynamics. Polyploidy, or whole-genome duplication (WGD), represents one of the most significant complicating factors, with all angiosperms sharing ancestral polyploid events and approximately 24% of existing plant species being recent polyploids [12]. These duplication events create massive genetic redundancy that allows for functional diversification between duplicates, leading to complex gene families that resist simple orthology classification [12]. For example, studies of the GRAS transcription factor family across 15 representative plant species revealed that widespread expansion of GRASs was predominantly driven by polyploidization events rather than small-scale duplications [12]. This pattern is consistent across many transcription factor families in plants, whose evolution and expansion are frequently associated with WGD events, subsequently enhancing plant resistance to environmental stress [12].
Limitations of Traditional Methods in Plant Genomics
Traditional orthology inference methods developed for animal genomes, where one-to-one orthology is more prevalent, often perform poorly when applied to plant genomes. The bidirectional best hit approach, commonly used for one-to-one ortholog identification, has been shown to miss many orthologs in duplication-rich clades such as plants [15]. This limitation arises because gene duplications create complex relationships that cannot be adequately captured by simple pairwise comparisons. Additionally, methods that rely solely on sequence similarity scores without accounting for gene length bias demonstrate significant performance variations, with short sequences suffering from low recall rates (missing genuine orthologs) and long sequences suffering from low precision (incorrect ortholog assignments) [10]. These methodological limitations are particularly problematic in plant genomics, where gene families frequently exhibit substantial length variation and complex evolutionary histories shaped by repeated duplication events.
Performance Comparison: Orthogroups vs. One-to-One Orthologs
Benchmarking Results on Standardized Datasets
Comprehensive benchmarking studies provide empirical evidence for evaluating the performance of different orthology inference methods. The Quest for Orthologs initiative maintains community standards and benchmark datasets that enable objective comparisons. When tested on the 2011_04 benchmark dataset, OrthoFinder (representing the orthogroup approach) demonstrated 3-24% higher accuracy on the SwissTree test and 2-30% higher accuracy on the TreeFam-A test compared to all other methods, including those focused on one-to-one ortholog identification [13]. These tests assess ortholog inference accuracy against gold-standard trees, measuring precision, recall, and F-score. The following table summarizes key performance metrics from published benchmarks:
Table 1: Performance Comparison of Orthology Inference Methods
| Method | Approach | SwissTree F-Score | TreeFam-A F-Score | Plant WGD Handling | Gene Length Bias |
|---|---|---|---|---|---|
| OrthoFinder | Phylogenetic Orthogroups | 3-24% higher than alternatives [13] | 2-30% higher than alternatives [13] | Explicit gene tree reconciliation [13] | Corrected via normalization [10] |
| OMA | Pairwise Orthologs & HOGs | Not specified in results | Not specified in results | HOGs at taxonomic levels [11] | Not explicitly addressed |
| OrthoMCL | Graph-based Clustering | Lower performance [10] | Lower performance [10] | No special handling | Strong bias observed [10] |
| InParanoid | Pairwise Orthologs | Not specified in results | Not specified in results | Limited capability | Not explicitly addressed |
Handling of Plant-Specific Complexities
The superior performance of phylogenetic orthogroup methods in plant genomics stems from their inherent ability to address plant-specific challenges. By inferring rooted gene trees for all orthogroups and reconciling them with a rooted species tree, OrthoFinder can accurately distinguish orthologs from paralogs even in complex scenarios involving WGDs [13]. This capability is particularly valuable for studying plant transcription factor families, such as the GRAS family, where OrthoFinder analysis has revealed 6.9 million previously unobserved relationships through complete classification of these gene families in plants [10]. In contrast, methods focused on identifying one-to-one orthologs necessarily exclude genes with complex duplication histories, potentially discarding biologically significant relationships that are central to plant evolution and adaptation. The phylogenetic approach also enables the mapping of gene duplication events to specific branches in the species tree, providing critical insights into the evolutionary timing of WGD events and their functional consequences [13] [12].
Experimental Protocols for Orthology Analysis in Plants
Standard Workflow for Orthogroup Inference with OrthoFinder
For researchers investigating orthology relationships in plant species, following established protocols ensures robust and reproducible results. A standard OrthoFinder analysis begins with protein sequence preparation, requiring one FASTA file per species containing predicted protein sequences [14]. The basic execution command is straightforward: orthofinder -f /path/to/protein/fasta/files/ [14]. OrthoFinder then performs automated sequence similarity searching using DIAMOND (default) or BLAST, followed by orthogroup inference using length-normalized similarity scores to eliminate gene length bias [10]. The algorithm subsequently infers gene trees for each orthogroup using DendroBLAST, infers the rooted species tree from these gene trees, and finally identifies orthologs and gene duplication events through duplication-loss-coalescence analysis of the rooted gene trees [13]. This comprehensive workflow produces a complete set of results including orthogroups, orthologs, gene trees, the species tree, and gene duplication events mapped to both gene and species trees [13].
Specialized Protocol for Polyploid Plant Species
When analyzing polyploid plant species or plant groups with known WGD events, modified protocols enhance orthology inference accuracy. The analysis should include outgroup species to improve rooting accuracy for gene trees, which subsequently increases orthogroup inference accuracy by up to 20% according to Orthobench benchmarks [14]. For large-scale analyses across dozens of species, the --assign option in OrthoFinder version 3.0 enables efficient addition of new species to existing orthogroups without recomputing the entire analysis [14]. Researchers should leverage the Hierarchical Orthogroups output (N0.tsv, N1.tsv, etc.) rather than the deprecated Orthogroups.tsv file, as these provide more accurate orthogroup definitions at each level of the species tree [14]. For plant groups with known WGD events, the resulting gene trees should be explicitly examined for concentrations of gene duplication events at specific species tree branches, as these often correspond to historical polyploidization events [13] [12].
Experimental Validation of Orthology Predictions
Computational predictions of orthology require experimental validation, particularly for plant species with complex genomes. Systematic biological integration across genomics, transcriptomics, metabolomics, and phenomics provides robust validation, as demonstrated in studies of GRAS transcription factors across 15 plant species [12]. For example, orthology relationships inferred for polyploidy-related Chenopodium quinoa GRASs (CqGRASs) were validated through integration with flavonoid pathway analysis, protein interaction mapping, and examination of population transcriptomes from the 1000 Plants (OneKP) project [12]. This integrated approach confirmed that GRASs interact with auxin and photosynthetic pathways to regulate flavonoid biosynthesis, enabling plants to adapt to environmental stress [12]. Such validation strategies are particularly important for orthology predictions in plant species, where functional conservation may persist despite complex gene family expansions through polyploidization.
Essential Research Toolkit for Plant Orthology Analysis
Table 2: Essential Tools and Resources for Plant Orthology Research
| Tool/Resource | Function | Application in Plant Research |
|---|---|---|
| OrthoFinder | Phylogenetic orthogroup inference | Comprehensive orthology, gene tree, species tree, and duplication inference [13] [14] |
| OMA Browser | Pairwise orthologs and HOGs | Identification of one-to-one orthologs and hierarchical groups [11] |
| Diamond | Accelerated sequence similarity | Fast all-vs-all protein sequence comparisons [13] |
| Plant Orthology Browser | Synteny and orthology visualization | Interactive exploration of gene order and orthology across 20 plant species [15] |
| PhylomeDB | Phylogenetic tree database | Exploration of evolutionary histories of plant genes [13] |
| OneKP Database | Plant transcriptome resource | Validation across 1000 plant transcriptomes [12] |
The choice between orthogroup and one-to-one ortholog approaches in plant research should be guided by specific biological questions and the genomic complexities of the study system. For investigations of gene family evolution, polyploidy impacts, and comparative genomics across deep evolutionary timescales, orthogroup methods (particularly phylogenetic approaches like OrthoFinder) provide superior accuracy and biological insights. The empirical benchmarking data clearly demonstrates their advantage in handling plant-specific challenges like WGD and gene length bias. Conversely, for studies focused on specific pairwise species comparisons where one-to-one orthology is expected to be prevalent, methods like OMA may offer sufficient resolution with potentially simpler interpretation. As plant genomics continues to expand with more sequenced genomes, phylogenetic orthogroup methods represent the most robust framework for unraveling the complex evolutionary history of plant genes and genomes, ultimately enabling discoveries in plant biology, breeding, and biotechnology.
The Impact of Whole-Genome Duplication on Plant Orthogroup Evolution
Whole-genome duplication (WGD) is a transformative evolutionary event that has profoundly shaped the genomic architecture of flowering plants. These episodes of polyploidization provide the raw genetic material for evolutionary innovation by simultaneously duplicating every gene in the genome. The subsequent fate of these duplicates—through retention, functional diversification, or loss—fundamentally influences orthogroup composition and dynamics across plant lineages. Orthogroups, defined as sets of genes descended from a single gene in the last common ancestor of the species being compared, provide a critical framework for understanding gene family evolution across deep phylogenetic distances. This review examines how WGD events have impacted orthogroup evolution in plants, synthesizing recent advances in comparative genomics and phylogenomics to elucidate the patterns, mechanisms, and functional consequences of duplicate gene retention and loss.
The prevalence of WGD in plant evolution is striking—all extant angiosperms are in fact ancient polyploids that have undergone at least two separate WGDs [16]. Subsequent and sometimes repeated WGDs have been reported in all major clades, making plants an exceptional system for studying the long-term evolutionary consequences of polyploidization [16]. Recent studies have revealed that WGDs are not randomly distributed through time but often cluster around periods of environmental upheaval, such as the Cretaceous-Paleogene (K-Pg) extinction event approximately 66 million years ago [16]. This temporal association suggests that the genomic plasticity afforded by WGD may enhance survival capacity during times of ecological stress, with lasting implications for orthogroup evolution and functional diversification.
The Genomic and Evolutionary Context of WGD in Plants
Prevalence and Timing of Plant WGD Events
Plant genomes have experienced multiple cycles of polyploidization throughout their evolutionary history. Genomic analyses indicate that 50–70% of angiosperms have experienced one or more episodes of chromosome doubling in their evolutionary trajectory [17]. Bayesian evolutionary analyses incorporating 38 full genome sequences and three transcriptome assemblies demonstrate a strongly nonrandom pattern of genome duplications over time, with significant clustering around the K-Pg boundary [16]. This period of mass extinction likely presented ecological conditions that favored the establishment of polyploid lineages, potentially through increased genetic buffering and phenotypic plasticity.
Beyond this catastrophic boundary, successive waves of WGD have occurred in various plant lineages. Table 1 summarizes major WGD events across plant taxa and their associated evolutionary implications. The Poaceae family (grasses), for instance, experienced several ancient polyploidizations, including the tau WGD shared by most monocots, the sigma triplication shared by the order Poales, and the Poaceae-specific rho WGD [18]. More recent lineage-specific WGDs have been identified in subfamilies including Bambusoideae, Pooideae, Panicoideae, Chloridoideae, and Oryzoideae [18].
Table 1: Major Whole-Genome Duplication Events in Plant Lineages
| Taxonomic Group | WGD Event(s) | Evolutionary Implications | Key References |
|---|---|---|---|
| Angiosperms (general) | Multiple ancient WGDs, often clustering around K-Pg boundary (~66 mya) | Genomic plasticity during environmental stress; basis for all extant angiosperms being paleopolyploids | [16] |
| Poaceae (grasses) | tau (shared by monocots), sigma (Poales), rho (Poaceae-specific), plus subfamily-specific WGDs | Differential duplicate retention linked to environmental adaptations (C4 photosynthesis, cold tolerance, aquatic growth) | [18] |
| Vertebrates | Two rounds at vertebrate origin (~500-550 mya) | Increased network complexity; enrichment of transcription factors and developmental genes | [19] |
| Arabidopsis thaliana | Alpha and beta duplications | Model for studying duplicate gene retention and functional divergence | [16] |
Contrasting Evolutionary Fates of Polyploids
The evolutionary trajectory following WGD remains a subject of intense debate, with evidence supporting two contrasting perspectives. One view characterizes polyploidy as an evolutionary dead end, citing the numerous immediate challenges faced by newly formed polyploids. These include meiotic and mitotic abnormalities from improper pairing of subgenomes during cell division, resulting in genomic instability that detrimentally affects fertility and fitness [16]. Additionally, newly formed polyploids face the "minority cytotype disadvantage," a frequency-dependent reproductive barrier that occurs when unreduced 2n gametes cross with reduced n gametes from diploid progenitors, producing less fit triploid hybrids [16]. These factors likely contribute to the observed lower speciation rates and higher extinction rates of polyploid plants compared to their diploid relatives.
The alternative perspective views polyploidy as a road toward evolutionary success, evidenced by the fact that all extant angiosperms and vertebrates are ancient polyploids [16]. An estimated 15% of flowering plant speciations involved ploidy increase, with this figure rising to 31% in ferns [16]. The evolutionary potential of polyploids stems from their duplicated genome, which provides thousands of novel genes for evolutionary experimentation. Although most duplicates are lost through pseudogenization, the retained fraction can lead to novel functionalities through mechanisms including neofunctionalization, subfunctionalization, and dosage effects [16]. Additionally, duplicates retained under dosage-balance constraints provide polyploids with a genetic "toolbox" that can be rewired to address new ecological challenges [16].
Methodological Framework for Orthogroup Analysis Post-WGD
Orthogroup Inference and WGD Detection
The accurate identification of orthogroups in the context of WGD requires specialized methodologies that account for the complex gene relationships resulting from polyploidization. OrthoFinder has emerged as a powerful tool for inferring orthogroups across multiple species, enabling researchers to distinguish between orthologs and paralogs in WGD-derived genomes [5]. This approach forms the foundation for comparative analyses that reveal patterns of gene retention and loss following polyploid events.
WGD detection itself relies on multiple lines of evidence. Synteny-based methods identify collinear blocks of duplicated genes within genomes, revealing the chromosomal scale of duplication events [18]. Phylogenomic approaches map gene duplications to specific nodes in species trees by comparing gene phylogenies with species trees [18]. Molecular dating of paralogous pairs using synonymous substitution rates (Ks) provides additional evidence, with Ks peaks indicating periods of widespread duplication [18]. The integration of these complementary methods has revealed previously unknown WGDs; a recent phylogenomic analysis of 363 grasses covering all 12 subfamilies reported nine previously unknown WGD events in addition to the established rho event [18].
The Orthologous Marker Groups (OMG) Method for Cross-Species Comparison
Recent methodological innovations have enhanced our ability to track orthogroup evolution across species. The Orthologous Marker Groups (OMG) method represents a significant advance for identifying cell types and comparing gene expression patterns across diverse plant species, overcoming challenges posed by frequent tandem duplications and WGDs in plants [5]. This approach identifies orthologous marker groups that can determine cell-type identity by counting overlapping orthologous gene groups between a query species and reference single-cell maps, employing statistical tests to quantify similarities between cell clusters.
The OMG method operates through a three-step process:
- Identification of top marker genes (typically N=200) for each cell cluster in each species using established approaches like Seurat
- Generation of orthologous gene groups for multiple plant species using OrthoFinder
- Pairwise comparisons using overlapping OMGs between clusters in query and reference species, with statistical evaluation via Fisher's exact test
This method successfully identified significant conservation in shared cell-type markers across monocots and dicots, revealing 14 dominant groups with substantial conservation [5]. Unlike integration-based methods that require extensive computational resources and can produce clusters with mixed cell identities, the OMG approach does not require cross-species data integration while still accurately determining inter-species cellular similarities [5].
Table 2: Key Methodological Approaches for Studying Orthogroup Evolution Post-WGD
| Method | Primary Application | Advantages | Limitations |
|---|---|---|---|
| Synteny Analysis | Identifying collinear blocks of duplicated genes | Strong evidence for WGD; reveals chromosomal architecture | Signal erodes with time due to rearrangements |
| Ks Distribution Analysis | Dating WGD events through synonymous substitution rates | Provides relative timing of duplication events | Can be confounded by multiple events; rate variation |
| Phylogenomic Profiling | Mapping GDs to species trees using gene tree-species tree comparisons | Identifies WGDs even without clear synteny; works with transcriptome data | Computationally intensive; requires careful curation |
| Orthologous Marker Groups (OMG) | Cross-species cell type identification and comparison | Accounts for gene family expansion; no integration needed | Requires reference datasets; optimized marker number varies |
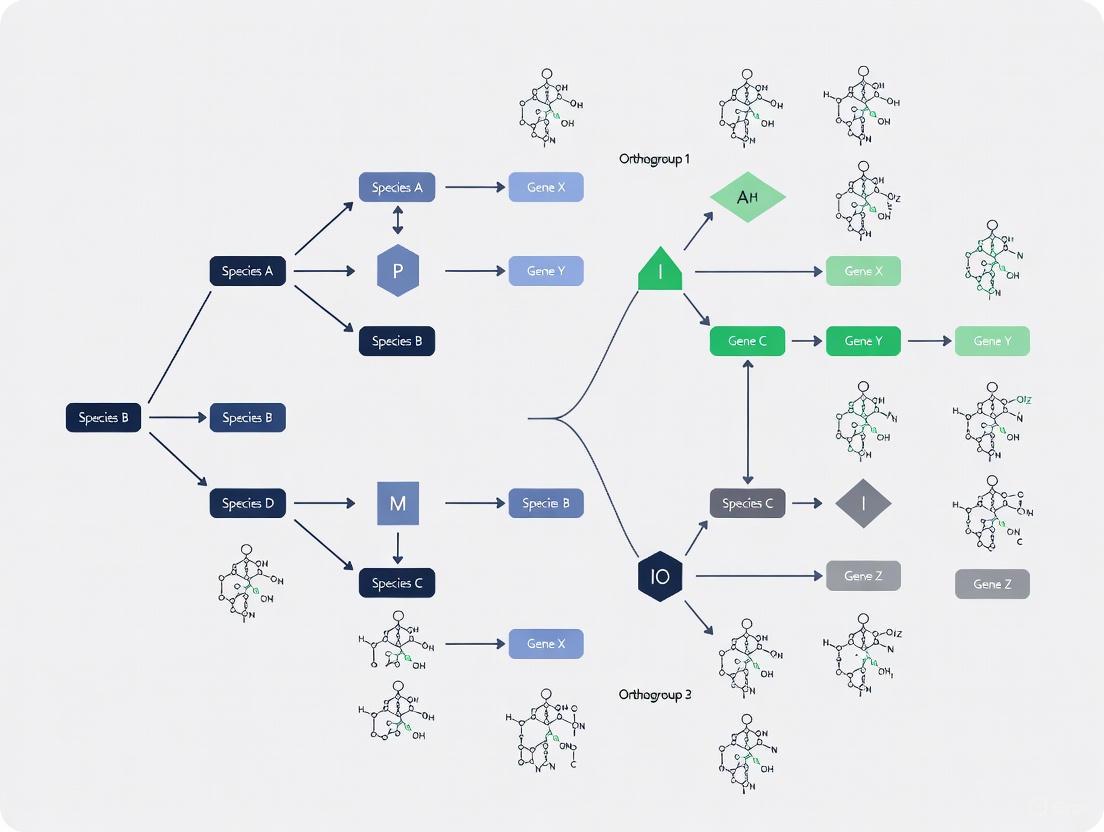
Experimental Workflow for Orthogroup Conservation Analysis
The following diagram illustrates a comprehensive experimental workflow for analyzing orthogroup conservation across species following WGD events:
Diagram 1: Experimental workflow for orthogroup conservation analysis across plant species following WGD events.
Differential Duplicate Retention and Loss Following WGD
Lineage-Specific Patterns of Gene Retention
One of the most significant insights from recent comparative genomic studies is the phenomenon of differential duplicate retention across lineages following shared WGD events. Rather than random patterns of gene loss, systematic biases emerge where certain gene families and functional categories are preferentially retained in specific lineages, potentially contributing to adaptive evolution.
In Poaceae, the rho WGD exemplifies this pattern. A phylogenomic analysis of 363 grasses revealed that rho-derived duplicates show differential retention among subfamilies, with specific retentions linked to environmental adaptations [18]. For instance:
- ACOT duplicates were preferentially retained in Oryzoideae, potentially supporting adaptation to aquatic environments
- CK2β duplicates showed retention in Pooideae, possibly contributing to cold response mechanisms
- SPIRAL1 duplicates were maintained in Bambusoideae, potentially facilitating rapid cell elongation
- PAI1 duplicates persisted in Panicoideae, likely involved in drought and cold stress responses
This lineage-specific retention pattern extends beyond Poaceae. Analysis of yeast species that shared an ancient WGD revealed that differential loss of duplicates created situations where 4-7% of single-copy genes between any two species were actually paralogs rather than orthologs [18]. This phenomenon complicates orthogroup inference and highlights how WGD can create evolutionary trajectories that differ across lineages even when they share a common polyploid ancestor.
Functional Consequences of Differential Retention
The functional implications of differential duplicate retention are profound, influencing morphological evolution, physiological adaptations, and ecological specialization. Several case studies illustrate how retained duplicates from WGD events have been co-opted for novel functions:
In rice, the rho-derived paralogs MADS50 and MADS51 act upstream of the Early heading date1 gene to regulate flowering transition but are differentially regulated by histone methylation [18]. This represents a case of subfunctionalization where the ancestral function has been partitioned between duplicates, allowing for more complex regulation of a key developmental process.
Another compelling example comes from rice SD1 genes involved in gibberellin biosynthesis. One copy (LOC_Os01g66100) promotes internode elongation in plants grown in deep-water, while its duplicate does not share this function [18]. This represents neofunctionalization directly linked to adaptation to specific environmental conditions—in this case, periodic flooding.
In the vertebrate lineage, the two rounds of WGD approximately 500 million years ago significantly increased the complexity of regulatory networks. WGD-derived transcription factors play a prominent role in maintaining strong regulatory redundancy, and complex network motifs such as combinations of feed-forward loops and bifan arrays are specifically enriched in the human regulatory network due to WGD events [19]. This enhanced combinatorial organization potentially increases network robustness and enables sophisticated functions like signal integration and noise control.
Research Reagent Solutions for Orthogroup Analysis
Table 3: Essential Research Reagents and Resources for Orthogroup Conservation Studies
| Reagent/Resource | Primary Function | Application Examples | Key Considerations |
|---|---|---|---|
| OrthoFinder | Infers orthogroups from protein sequences | Identifying orthogroups across multiple species; distinguishing orthologs/paralogs | Handles large datasets; accounts for gene duplication events |
| BEAST Software | Bayesian evolutionary analysis | Molecular dating of WGD events; incorporating fossil uncertainty | Computationally intensive; allows relaxed clock models |
| 3D-GDP Database | Plant 3D genome database | Comparing 3D genome structures across species; identifying conserved TADs | Includes 26 plant species; enables evolutionary comparisons |
| OHNOLOGS Database | Catalog of WGD-derived gene pairs | Identifying vertebrate WGD pairs; analyzing ohnolog retention patterns | High-confidence list of paralogues; updated gene names |
| Seurat | Single-cell RNA-seq analysis | Identifying marker genes for cell types; data normalization and clustering | Optimized parameters needed for plant data |
| SynMap | Synteny analysis and visualization | Identifying collinear blocks; detecting WGD through synteny | Signal erosion for ancient WGDs; requires genome annotations |
Duplicate Retention Patterns and Functional Implications
The fate of duplicated genes following WGD is nonrandom, with specific functional categories showing preferential retention across lineages. The following diagram illustrates key processes in duplicate retention and functional evolution:
Diagram 2: Post-WGD gene retention mechanisms and their evolutionary outcomes.
Dosage-Sensitive Gene Retention
Dosage-balance constraints play a crucial role in determining which duplicates are retained following WGD. Genes encoding components of multiprotein complexes or those involved in dose-sensitive regulatory pathways are preferentially maintained because altering their stoichiometry would disrupt essential cellular functions [16]. This dosage-sensitive retention is particularly evident for transcription factors and signaling components, which are consistently overrepresented among WGD-derived duplicates compared to small-scale duplicates [19].
In plants, duplicates retained through dosage constraints include many regulatory and developmental genes guarded against loss through dosage-balance constraints on the stoichiometry of duplicated pathways and macromolecular complexes [16]. The resolution of these constraints over time provides polyploid species with a genetic toolkit that can be rewired to execute novel functionality, potentially enabling response to new ecological opportunities and challenges [16].
Comparative Analysis of Retention Patterns
Systematic comparisons of duplicate retention patterns across angiosperms reveal both conserved and lineage-specific trends. A phylogenomic analysis of 41 plant species found that WGDs dating to the K-Pg boundary were followed by nonrandom retention of genes potentially involved in stress response and environmental adaptation [16]. This pattern suggests that polyploid establishment may be promoted during times of environmental stress, with lasting impacts on orthogroup composition.
In vertebrates, WGD-derived genes are threefold more likely than non-WGD genes to be involved in cancers and autosomal dominant diseases [19]. This observation suggests that WGD genes are intrinsically "dangerous" in that they are more susceptible to dominant deleterious mutations, possibly because they often encode highly connected proteins in regulatory networks. Despite this potential danger, WGD genes are more frequently involved in signaling, development, and transcriptional regulation and are enriched in Gene Ontology categories associated with organismal complexity [19].
Whole-genome duplication represents a fundamental evolutionary force that has repeatedly shaped plant genome architecture and orthogroup composition. The integration of comparative genomics, phylogenomics, and functional genetics has revealed complex patterns of duplicate gene retention and loss that extend far beyond random fractionation. Instead, differential retention following WGD follows predictable patterns influenced by dosage sensitivity, functional category, and ecological context.
The evidence reviewed herein demonstrates that WGD provides raw genetic material for evolutionary innovation through several non-mutually exclusive mechanisms: preservation of dosage-sensitive regulators, subfunctionalization of developmental pathways, neofunctionalization in response to environmental challenges, and rewiring of regulatory networks. These processes collectively contribute to the evolutionary success of polyploid lineages and their adaptive diversification across ecological gradients.
Future research in plant orthogroup evolution will benefit from increased taxonomic sampling across key phylogenetic nodes, enhanced computational methods for distinguishing orthologs and paralogs in polyploid genomes, and functional validation of WGD-derived genes implicated in adaptive evolution. As genomic resources continue to expand across the plant tree of life, so too will our understanding of how whole-genome duplications have sculpted orthogroup evolution and contributed to the remarkable diversity of flowering plants.
Orthology research is fundamental to comparative genomics, enabling scientists to trace the evolutionary history of genes across different species. In plant sciences, this is particularly complex and powerful due to frequent gene duplication events and whole-genome duplications that shape plant genomes. Resources for plant orthology research provide the critical foundation for identifying functional equivalents across species, transferring gene functional annotations, and studying the evolution of traits. This guide objectively compares several key databases and computational tools—OrthoFinder, the OMG Browser, AgroLD, and 3D-GDP—focusing on their specific applications, data content, and performance in orthogroup conservation analysis across plant species.
The following table summarizes the core features and intended use cases for each major resource, providing a baseline for comparison.
Table 1: Key Databases and Tools for Plant Orthology Research
| Resource Name | Primary Function | Key Inputs | Key Outputs | Scope (Number of Species) | Unique Strength / Focus |
|---|---|---|---|---|---|
| OrthoFinder [10] | Orthogroup Inference & Orthology Prediction | Protein sequence files from multiple species | Orthogroups, Gene Trees, Orthologues | Highly scalable (1,000s) | Algorithmic accuracy; solves gene-length bias in inference |
| OMG Browser [5] | Cell Type Identification & Conservation | scRNA-seq data and cluster marker genes | Cell type annotations based on orthologous markers | 15 plant species | Cross-species cell identity conservation from single-cell data |
| AgroLD [20] | Integrated Knowledge Graph for Hypothesis Generation | N/A (Pre-integrated data from >150 sources) | Consolidated gene, protein, pathway, and phenotype data | 51 plant species | Data integration and exploration via semantic web technologies |
| 3D-GDP [21] | 3D Genome Structure Comparison & Conservation | N/A (Pre-processed Hi-C and genomic data) | Conserved TADs, loops, compartments, and 3D structures | 26 plant species | Conservation of 3D genome structures and their regulatory impact |
Performance and Experimental Data
Orthogroup Inference Accuracy: OrthoFinder
OrthoFinder is a foundational algorithm for inferring orthogroups from protein sequences. Its performance is benchmarked against other methods using real manually-curated datasets [10].
Table 2: OrthoFinder Performance Benchmarking on OrthoBench Dataset
| Performance Metric | OrthoMCL (Comparison) | OrthoFinder | Improvement |
|---|---|---|---|
| Overall Accuracy (F-score) | Not explicitly stated (Baseline) | 8% to 33% more accurate [10] | +8% to +33% |
| Precision | Low for long genes [10] | High across all gene lengths [10] | Substantially increased |
| Recall | Low for short genes [10] | No dependency on gene length [10] | Dramatically improved for short genes |
| Key Innovation | Uses raw BLAST scores | Uses novel length-normalized BLAST scores [10] | Eliminates gene-length bias |
Cell Type Conservation Mapping: OMG Browser
The Orthologous Marker Gene Groups (OMG) method addresses the challenge of identifying conserved cell types across plant species using single-cell RNA sequencing (scRNA-seq) data. Its performance was validated through several key experiments [5].
Table 3: Experimental Validation of the OMG Method
| Experiment / Test | Species Compared | Key Performance Result | Experimental Context |
|---|---|---|---|
| Dicot Root Comparison | Tomato (query) vs. Arabidopsis (reference) | 12/13 tomato cell clusters showed exact or functional matches to Arabidopsis [5] | 165 cluster pairs tested; 24 showed significant shared OMGs (FDR < 0.01) [5] |
| Monocot-Dicot Comparison | Rice (query) vs. Arabidopsis (reference) | Identified 14 conserved cell cluster pairs (FDR < 0.01) [5] | One-to-one ortholog mapping only identified 8 pairs, with lower accuracy [5] |
| Large-Scale Mapping | 1 million cells, 268 clusters across 15 species [5] | Revealed 14 dominant groups with conserved cell-type markers [5] | Demonstrates scalability and conservation across monocots and dicots |
Detailed Experimental Protocols
Protocol: Orthologous Marker Gene Groups (OMG) Analysis
This protocol is adapted from the methodology used to validate the OMG Browser, designed for identifying conserved cell types across species using scRNA-seq data [5].
Input Data Preparation:
- Single-Cell Data Clustering: Process your query scRNA-seq dataset through a standard analysis pipeline (e.g., Seurat) to identify cell clusters [5].
- Marker Gene Identification: For each cell cluster in the query species, identify the top N marker genes (N=200 was used in the original study). Using a consistent N is critical for statistical comparability [5].
Orthologous Group Mapping:
- Orthogroup Assignment: Map the marker genes from all species involved (query and references) to pre-computed orthogroups. The original study used OrthoFinder to generate orthologous groups for 15 plant species [5].
Cross-Species Comparison:
- Overlap Calculation: For every pair of clusters (query cluster vs. reference cluster), count the number of Orthologous Marker Gene Groups (OMGs) they share.
- Statistical Significance Testing: Perform a Fisher's exact test for each query-reference cluster pair to determine if the number of shared OMGs is significantly greater than expected by chance. Correct for multiple testing (e.g., using FDR).
Cell Type Annotation:
- Assignment: Annotate cell types in the query species by transferring labels from reference clusters (e.g., from Arabidopsis) with which they share a significant number of OMGs.
- Validation: Where possible, validate annotations using independent data, such as promoter-GFP lines as done in the tomato validation [5].
Protocol: Orthogroup Inference with OrthoFinder
This protocol outlines the core steps for using OrthoFinder to infer orthogroups, which is also a key component of the OMG method [5] [10].
Input: Gather protein sequence files (in FASTA format) for all species to be analyzed.
Sequence Similarity Search: OrthoFinder performs an all-vs-all BLAST search of the protein sequences. The innovative step is the normalization of BLAST bit scores to eliminate gene length and phylogenetic distance bias [10].
Graph Construction and Clustering: The normalized scores are used to construct a graph where nodes are genes and edges represent sequence similarity. The MCL algorithm is then used to cluster this graph into orthogroups [10].
Output Analysis: OrthoFinder generates comprehensive results, including:
- The list of orthogroups and their member genes.
- Gene trees and rooted species tree.
- Matrices of orthologue counts between species.
- Comparative statistics about the analysis.
Workflow and Resource Selection Diagrams
Figure 1: A workflow to guide the selection of the most appropriate resource based on the researcher's specific goal.
Figure 2: The core analytical workflow of the OMG method for cross-species cell type identification.
Table 4: Essential Computational Tools and Data Sources for Plant Orthology Research
| Tool / Resource | Category | Primary Function in Orthology Research | Key Application Note |
|---|---|---|---|
| OrthoFinder [5] [10] | Core Algorithm | Infers orthogroups from protein sequences; foundational for OMG groups. | Provides the evolutionary groups for downstream comparative analyses. |
| Single-Cell RNA-seq Data [5] | Input Data | Enables identification of cell-type-specific marker genes for comparison. | High-quality clustering is a prerequisite for accurate OMG analysis. |
| OrthoBench [10] | Benchmarking Dataset | A manually curated set of orthogroups for validating inference methods. | Used to demonstrate OrthoFinder's accuracy improvements (8-33%) [10]. |
| Seurat [5] | Software Pipeline | Standard tool for scRNA-seq data analysis, including clustering and marker gene finding. | Used in the OMG protocol to generate the initial input data [5]. |
| Fisher's Exact Test | Statistical Method | Determines if shared OMGs between clusters are statistically significant. | Critical for controlling false discoveries in cross-species mapping [5]. |
In comparative genomics, an orthogroup is defined as the set of genes that descended from a single gene in the last common ancestor of all species being considered, encompassing both orthologs and paralogs [10]. This conceptual framework provides a powerful foundation for understanding evolutionary relationships and functional conservation across species. The identification and analysis of orthogroups have become fundamental to biological research, offering a coherent framework for extrapolating biological knowledge between organisms and illuminating the evolutionary history of gene families [10].
The study of orthogroups is particularly valuable in plant evolutionary biology, where gene duplication events—including whole-genome duplications and small-scale duplications—have been major drivers of gene family evolution and diversification [22]. By tracing the evolutionary trajectories of orthogroups across plant species, researchers can distinguish conserved core genes from lineage-specific innovations, revealing how evolutionary pressures shape gene function and biological processes [23] [24]. This approach has revealed millions of previously unobserved evolutionary relationships and provided insights into how plants adapt to environmental challenges and pathogens [10] [22].
Orthogroup Inference Methodologies: A Comparative Analysis
Software Tools for Orthogroup Inference
Various computational methods have been developed to infer orthogroups from genomic data, each employing distinct algorithms and offering different performance characteristics. The table below summarizes the key tools available for orthogroup inference:
Table 1: Comparison of Orthogroup Inference Software Tools
| Software | Algorithm Basis | Key Features | Strengths | Applicability |
|---|---|---|---|---|
| OrthoFinder | Phylogenetic orthology inference | Infers gene trees, rooted species trees, gene duplication events | Highest ortholog inference accuracy (3-30% better than others) [13] | Whole-genome comparisons across hundreds of species |
| OrthoQuantum | Binary phylogenetic profiling | Visualization of phylogenetic profiles, correlation analysis | Specialized for eukaryotic proteins, interactive visualization [23] | Analysis of co-inherited proteins and lineage-specific innovations |
| OrthoMCL | Markov Cluster algorithm | Graph-based clustering of BLAST scores | Widely used, extensive legacy citation record [10] | General-purpose orthogroup identification |
| InParanoid | Pairwise orthology detection | Focus on orthologs and paralogs between two species | High precision for pairwise comparisons [23] | Two-species comparative analyses |
| OMA | Pairwise and groupwise inference | Identifies orthologs and orthogroups using evolutionary distances | Handles complex orthology relationships [10] | Multi-species comparative genomics |
Quantitative Performance Comparison
Independent benchmarking studies have provided quantitative assessments of orthogroup inference methods. The most comprehensive evaluation comes from the Quest for Orthologs benchmarking service, which assesses performance using manually curated reference datasets:
Table 2: Performance Metrics of Orthogroup Inference Methods on Quest for Orthologs Benchmarks
| Method | SwissTree F-Score | TreeFam-A F-Score | Species Tree Discordance (pseudo-F-score) | Gene Length Bias | Computational Speed |
|---|---|---|---|---|---|
| OrthoFinder | 0.873 [13] | 0.839 [13] | 0.791 [13] | Minimal [10] | Fast (uses DIAMOND) [13] |
| OrthoMCL | 0.67 (estimated) | 0.645 (estimated) | 0.61 (estimated) | Significant [10] | Moderate |
| OMA | 0.72 (estimated) | 0.70 (estimated) | 0.68 (estimated) | Moderate | Slow |
| InParanoid | 0.71 (estimated) | 0.69 (estimated) | 0.65 (estimated) | Low | Fast for pairwise |
OrthoFinder demonstrates superior performance across multiple metrics, with benchmark results showing it is 3-24% more accurate on SwissTree and 2-30% more accurate on TreeFam-A compared to other methods [13]. This performance advantage stems from its phylogenetic approach and the solution to fundamental biases in whole-genome comparisons, particularly gene length bias that significantly affects other methods like OrthoMCL [10].
Experimental Approaches for Orthogroup Analysis
Standard Orthogroup Inference Protocol
A typical workflow for orthogroup analysis involves sequential computational steps from sequence preparation to evolutionary interpretation:
Diagram 1: Orthogroup analysis workflow showing key computational stages.
Step 1: Data Collection and Preparation
- Obtain proteome sequences from public databases (NCBI, Phytozome, Plaza) or newly sequenced genomes [22]
- For plant studies, include reference sequences from well-annotated genomes (e.g., Arabidopsis thaliana) [25]
- Format sequences and create local databases for efficient searching
Step 2: Sequence Similarity Analysis
- Perform all-vs-all sequence similarity searches using tools like DIAMOND [13] or BLAST [10]
- Apply sequence length normalization to correct for inherent biases in similarity scores [10]
- Generate normalized scores for accurate orthogroup inference
Step 3: Orthogroup Clustering
- Apply clustering algorithms (MCL in OrthoFinder, alternative methods in other tools) to identify orthogroups [22]
- Define orthogroups as sets of genes descended from a single ancestral gene
- Validate clusters against known gene families or manually curated datasets
Advanced Phylogenetic Orthology Inference
Modern orthogroup analysis has evolved beyond simple clustering to incorporate sophisticated phylogenetic methods:
Diagram 2: Phylogenetic orthology inference methodology.
Phylogenetic Tree Construction
- Infer gene trees for each orthogroup using methods like DendroBLAST or maximum likelihood approaches [13]
- Reconstruct the rooted species tree from gene trees using statistical methods [13]
- Root individual gene trees using the species tree as a reference
Evolutionary Event Identification
- Map gene duplication events to specific locations on both gene trees and species trees
- Distinguish orthologs from paralogs using phylogenetic relationships rather than similarity scores
- Account for incomplete lineage sorting and other confounding evolutionary processes
Case Study: Conserved Plant Defense Mechanisms Revealed Through Orthogroup Analysis
Identification of Evolutionarily Conserved Nematode-Responsive Genes
A compelling application of orthogroup analysis in plant evolutionary biology comes from a study of resistance to root-knot nematodes (RKNs), which identified evolutionarily conserved defense genes across multiple plant species [24]. The experimental approach demonstrates how orthogroup analysis can reveal functional conservation:
Table 3: Experimental Design for Identifying Conserved Plant Defense Genes
| Research Component | Description | Species Utilized | Analysis Output |
|---|---|---|---|
| Comparative Genomics | Compared predicted proteomes of 22 plant species spanning 214 million years of evolution | 22 plant species including important crops [24] | 35,238 protein orthogroups identified, 6,132 universal across all species |
| Transcriptome Analysis | Analyzed RNA-seq data from RKN-resistant genotypes challenged by Meloidogyne spp. | Wild peanut, coffee, soybean, African rice [24] | 2,597-653 differentially expressed genes identified per species |
| Orthogroup Integration | Classified DEGs into previously identified orthogroups | Cross-species analysis | 17 orthogroups containing DEGs from all resistant genotypes |
| Functional Characterization | Annotated genes within conserved orthogroups | - | Genes related to signaling, secondary metabolites, cell wall, plant defense |
The research identified 17 orthogroups universally conserved across 22 plant species that contained differentially expressed genes during resistance response to nematode infection in all four resistant genotypes studied [24]. These orthogroups contained 364 genes involved in signaling, secondary metabolite production, cell wall-related functions, peptide transport, transcription regulation, and plant defense, revealing evolutionarily conserved immune responses to pathogen challenge [24].
Nucleotide-Binding Site (NBS) Gene Family Evolution in Plants
Another significant case study analyzed the diversification of nucleotide-binding site (NBS) domain genes, which encode disease resistance proteins in plants [22]. This research employed orthogroup analysis to understand the evolutionary history of this critical gene family:
Methodological Approach:
- Identified 12,820 NBS-domain-containing genes across 34 plant species from mosses to monocots and dicots [22]
- Classified genes into 168 classes based on domain architecture patterns
- Utilized OrthoFinder for orthogroup analysis, identifying 603 orthogroups with core and lineage-specific groups [22]
- Conducted expression profiling to validate functional relevance under biotic stress
Key Findings:
- Discovered both classical (NBS-LRR, TIR-NBS-LRR) and species-specific structural patterns
- Identified core orthogroups (OG0, OG1, OG2) conserved across multiple species and unique orthogroups specific to particular lineages
- Demonstrated tandem duplications as a key mechanism for NBS gene family expansion
- Validated functional importance through expression analysis showing upregulation of specific orthogroups (OG2, OG6, OG15) under biotic stress
This comprehensive analysis provided insights into how plants maintain extensive NLR repertoires and how these disease resistance genes evolve through duplication and diversification events [22].
Essential Research Reagents and Computational Tools
Successful orthogroup analysis requires a suite of bioinformatic tools and resources. The following table outlines key components of the orthogroup analysis toolkit:
Table 4: Essential Research Reagents and Computational Tools for Orthogroup Analysis
| Tool Category | Specific Tools | Function | Application Context |
|---|---|---|---|
| Sequence Databases | NCBI, Phytozome, Plaza, Ensemble | Source of proteome sequences and annotations | Data acquisition for cross-species comparisons [25] [22] |
| Sequence Search | DIAMOND, BLAST, HMMER | Identify sequence similarities and domain conservation | Initial homology detection and orthogroup inference [13] [22] |
| Orthogroup Inference | OrthoFinder, OrthoMCL, OrthoQuantum | Identify groups of orthologous genes | Core analysis identifying evolutionarily related genes [23] [13] [10] |
| Phylogenetic Analysis | MAFFT, FastTree, DendroBLAST | Multiple sequence alignment and tree inference | Evolutionary analysis and orthology delineation [13] [22] |
| Expression Analysis | DESeq, EdgeR | Differential expression analysis from RNA-seq | Functional validation of orthogroups in specific conditions [24] |
| Visualization | PhyD3, JavaScript-based interfaces | Visualize phylogenetic profiles and relationships | Interpretation and communication of results [23] |
Orthogroup analysis has emerged as an indispensable methodology for understanding functional conservation across species. The comparative evaluation presented here demonstrates that phylogenetic approaches like OrthoFinder provide significant advantages in accuracy compared to similarity score-based methods, with benchmarks showing 3-30% improvement in ortholog inference [13]. The case studies on plant defense genes illustrate how this approach can reveal evolutionarily conserved immune responses [24] and gene family diversification patterns [22].
For researchers investigating plant evolution and functional genomics, orthogroup analysis offers a powerful framework for identifying core conserved genes versus lineage-specific innovations. The integration of comparative genomics with transcriptomic data through orthogroup classification enables the discovery of biologically significant genes with conserved functions across deep evolutionary timescales. As genomic data continue to accumulate, orthogroup analysis will remain fundamental to extracting meaningful biological insights from sequence information and understanding the evolutionary principles that shape gene function across the plant kingdom.
Methodological Framework: From Orthology Inference to Functional Analysis
In the field of plant genomics, accurately identifying evolutionary relationships between genes across species is fundamental to research on trait evolution, gene function conservation, and genomic diversity. Orthology inference—distinguishing genes separated by speciation events (orthologs) from those separated by duplication events (paralogs)—provides the critical framework for these comparative analyses. Multiple computational methods have been developed to infer orthology, each employing different algorithmic strategies with varying implications for accuracy, scalability, and suitability for plant genomic studies, which often involve complex genomic histories including polyploidization events.
Among these methods, OrthoFinder has emerged as a leading tool that combines high accuracy with comprehensive phylogenetic analysis. This guide objectively compares OrthoFinder's performance against other established orthology inference methods, with a specific focus on applications in plant species research where orthogroup conservation analysis is paramount. We present summarized quantitative benchmarking data, detailed experimental protocols, and essential resource information to assist researchers in selecting and implementing orthology inference pipelines for their plant genomics studies.
Orthology inference methods generally fall into two categories: those that use graph-based clustering of sequence similarity scores (heuristic methods) and those that use phylogenetic tree-based approaches. OrthoFinder uniquely bridges these approaches by providing both fast graph-based orthogroup inference and sophisticated phylogenetic analysis in an integrated pipeline.
OrthoFinder's Phylogenetic Methodology
OrthoFinder implements a multi-step phylogenetic methodology that extends beyond simple sequence similarity comparisons [13]. The algorithm proceeds through several key stages:
- Orthogroup Inference: Initial grouping of genes into orthogroups using sequence similarity scores, with a normalization step that corrects for gene length bias—a significant source of error in orthogroup detection [10].
- Gene Tree Inference: Inference of gene trees for each orthogroup.
- Species Tree Inference: Analysis of gene trees to infer a rooted species tree.
- Gene Tree Rooting: Rooting of all gene trees using the rooted species tree.
- Orthology Inference: Duplication-loss-coalescence (DLC) analysis of rooted gene trees to identify orthologs, paralogs, and gene duplication events [13].
This comprehensive approach allows OrthoFinder to distinguish between orthologs and paralogs based on the phylogenetic history of genes rather than solely on sequence similarity, which can be confounded by variable evolutionary rates [13].
Competing Algorithms
Several other widely-used orthology inference methods provide alternative approaches:
- SonicParanoid: Utilizes machine learning to avoid unnecessary all-against-all alignments, focusing on speed and efficiency [26].
- Broccoli: Employs a clustering approach related to OrthoFinder but with different implementation strategies.
- OMA (Orthologous MAtrix): Uses all-against-all gene comparisons with Smith-Waterman to find homologous sequences and infers orthology relationships [26].
- FastOMA: A recently developed tool that leverages existing knowledge from the OMA database and uses a fast k-mer-based mapping approach to reference gene families before performing phylogenetic analysis [26].
- OrthoMCL: A traditional graph-based method that uses BLAST similarity scores and the MCL clustering algorithm, which suffers from gene length bias that OrthoFinder specifically addresses [10].
Performance Benchmarking in Plant Genomics
General Benchmarking on Standardized Tests
Independent benchmarking efforts through the Quest for Orthologs initiative provide comprehensive performance comparisons. On the SwissTree and TreeFam-A benchmarks—which assess accuracy against gold-standard trees—the default version of OrthoFinder demonstrated 3-24% and 2-30% higher accuracy, respectively, than any other method tested [13]. No single method was consistently second best to OrthoFinder, highlighting its unique positioning in the landscape of orthology inference tools.
Table 1: Orthology Inference Accuracy on Quest for Orthologs Benchmarks
| Method | SwissTree F-Score | TreeFam-A F-Score | Primary Methodology |
|---|---|---|---|
| OrthoFinder (Default) | Highest | Highest | Phylogenetic (Gene tree-based) |
| Other Methods (Best) | 3-24% lower | 2-30% lower | Varies (Graph-based, RBH, etc.) |
| OrthoFinder (Alternative configs) | +1-3% over default | +1-3% over default | Phylogenetic with enhanced settings |
Performance in Plant Species Research
A recent study specifically evaluated orthology inference algorithms in the context of plant genomes, analyzing eight Brassicaceae species in two groups: one comprising only diploids and another including diploids, two mesopolyploids, and one recent hexaploid genome [27]. This research is particularly relevant for plant genomics as it reflects the complex genomic histories common in plant lineages.
The study found that three algorithms—OrthoFinder, SonicParanoid, and Broccoli—were all helpful for initial orthology predictions, with orthogroup compositions reflecting the species' ploidy and genomic histories [27]. The diploid set showed a higher proportion of identical orthogroups across methods, while the diploid + higher ploidy set had a lower proportion of identically composed orthogroups, though the average degree of similarity between orthogroups was not different from the diploid set [27]. This suggests that while these three methods generally produce consistent results, the additional complexity introduced by polyploidization events leads to more frequent disagreements in orthogroup boundaries.
Experimental Protocols for Orthology Analysis
Standard OrthoFinder Workflow for Plant Genomes
The following workflow describes a typical OrthoFinder analysis suitable for plant genomic studies:
Proteome Acquisition and Preparation:
- Obtain protein sequences for all species of interest in FASTA format. For plant studies, sources include Ensembl Genomes (http://ensemblgenomes.org) and Phytozome (https://phytozome.jgi.doe.gov) [28].
- Use the longest transcript variant per gene when multiple isoforms exist. OrthoFinder provides a script for this with Ensembl proteomes.
- Use concise, informative species names in filenames (e.g., "A_thaliana.fa") as these will appear in result visualizations.
Running OrthoFinder:
- Basic command:
orthofinder -f [directory_with_fasta_files] - For large analyses or adding species to existing analysis, use the
--assignoption available in OrthoFinder 3.0+ [14].
- Basic command:
Results Exploration:
- Check the percentage of genes assigned to orthogroups (in
Comparative_Genomics_Statistics/Statistics_Overall.tsv). Values below 80% may indicate poor species sampling [29]. - Verify the inferred species tree (
Species_Tree/SpeciesTree_rooted.txt) matches biological expectations. - Identify orthologs of interest through the species-pair files in the
Orthologuesdirectory. - Examine gene trees (
Gene_Trees/orResolved_Gene_Trees/) for specific orthogroups of interest to confirm orthology/paralogy relationships.
- Check the percentage of genes assigned to orthogroups (in
Diagram 1: OrthoFinder analysis workflow
Species Selection Strategy for Plant Studies
The selection of species for analysis significantly impacts results, particularly in plant genomics with its frequent whole genome duplication events:
- Comparative analysis across a clade: Include all available species in the clade of interest. Generally avoid outgroups as they push back the point at which orthogroups are defined, reducing resolution [28].
- Identifying orthologs among few species: Include 6-10 species minimum to break up long branches in the species tree [28].
- Investigating specific evolutionary events: Include at least two species below the branch of interest, two species on the closest branch above, and two or more outgroup species [28].
Table 2: Key Research Reagent Solutions for Orthology Analysis
| Resource Type | Specific Examples | Function in Analysis |
|---|---|---|
| Proteome Data Sources | Ensembl Genomes, Phytozome, NCBI | Provides protein sequences in FASTA format for analysis [28] |
| Sequence Search Tools | DIAMOND, BLAST+, MMseqs2 | Performs rapid all-vs-all sequence comparisons [13] |
| Multiple Sequence Alignment | MAFFT, MUSCLE, Clustal Omega | Aligns sequences within orthogroups for tree inference |
| Phylogenetic Inference | FastTree, IQ-TREE, RAxML | Infers gene trees from sequence alignments |
| Visualization Tools | Dendroscope, ETE Toolkit, iTOL | Views and interprets species trees and gene trees [29] |
Discussion and Research Applications
Interpretation of Gene Duplication Events in Plants
A particular strength of OrthoFinder in plant genomics is its ability to identify and map gene duplication events to both gene trees and species trees. For example, when analyzing gene families expanded through whole genome duplication events common in plant evolution, OrthoFinder can distinguish between duplication events that occurred in common ancestors versus those that are lineage-specific [29]. The Gene_Duplication_Events directory in results provides files detailing these events, including measures of support based on retention of both duplicated copies in descendant species [29].
Hierarchical Orthogroups for Studying Plant Evolution
From version 2.4.0 onward, OrthoFinder infers Hierarchical Orthogroups (HOGs)—orthogroups defined at each node of the species tree, which are particularly valuable for studying plant evolution [14]. According to OrthoBench benchmarks, these phylogenetically-informed orthogroups are 12-20% more accurate than the graph-based orthogroups used by most other methods [14]. For plant researchers, this means HOGs can more accurately define gene families that diversified at specific points in plant evolutionary history, such as before or after the divergence of monocots and eudicots.
OrthoFinder provides a comprehensive platform for phylogenetic orthology inference that demonstrates leading accuracy on standardized benchmarks while offering specific advantages for plant genomics research. Its integrated phylogenetic approach, which infers rooted gene trees and species trees alongside orthogroups, enables researchers to accurately reconstruct complex evolutionary histories characteristic of plant genomes. While alternative methods like SonicParanoid and Broccoli show comparable performance in some plant-specific studies [27], OrthoFinder's unique combination of high accuracy, comprehensive phylogenetic analysis, and hierarchical orthogroup inference makes it particularly suited for investigating orthogroup conservation across plant species. As plant genomics continues to expand with more sequenced genomes, tools like OrthoFinder that can accurately resolve complex evolutionary relationships will remain essential for comparative genomic studies.
The rapid advancement of single-cell RNA sequencing (scRNA-seq) technology has revolutionized studies of cell identity and differentiation in plant biology [5]. However, a significant challenge persists in accurately identifying cell types across diverse plant species, particularly non-model organisms. This challenge stems primarily from the scarcity of known cell-type marker genes and the divergence of marker expression patterns between species [5]. Compounding this problem, frequent gene duplication events and whole-genome expansions in plants create complex gene families that complicate direct cross-species comparisons using simple one-to-one orthologs [5].
To address these limitations, a novel computational strategy termed Orthologous Marker Gene Groups (OMGs) has been developed. This approach leverages orthogroup conservation analysis to enable robust cell type identification across distantly related plant species without requiring computationally intensive cross-species data integration [5]. The OMG method represents a paradigm shift in comparative plant transcriptomics, moving beyond sequence-based orthology to incorporate functional conservation of cell-type specific expression patterns. This review provides a comprehensive comparison of the OMG approach against traditional methods, supported by experimental data and detailed methodologies.
Methodological Framework: How OMGs Work
Core Principles and Workflow
The OMG method operates on a fundamental insight: while individual marker genes may show divergent expression patterns across species, groups of orthologous genes often maintain conserved cell-type specificity [5]. This approach utilizes orthogroups—sets of genes descended from a single gene in the last common ancestor—as the unit of comparison rather than individual genes.
The implementation involves a structured workflow with three key stages:
- Marker Gene Identification: For each cell cluster in a scRNA-seq dataset, the top N marker genes (typically N=200) are identified using established tools like Seurat [5] [30].
- Orthogroup Construction: OrthoFinder or similar tools are used to generate orthologous gene groups across multiple plant species [5] [30].
- Statistical Comparison: Pairwise comparisons between clusters across species are performed using Fisher's exact test to identify significant overlaps in shared OMGs, with false discovery rate (FDR) correction for multiple testing [5].
Table: Key Parameters in the OMG Workflow
| Parameter | Typical Setting | Rationale | Impact of Variation |
|---|---|---|---|
| Number of Marker Genes (N) | 200 per cluster | Balances specificity and sensitivity | N<200: Rapid decrease in cross-species overlapsN>200: Reduced marker gene specificity |
| Statistical Threshold | FDR < 0.01 | Controls false discoveries in multiple testing | Less stringent: More matches but potential false positivesMore stringent: Potential missed valid matches |
| Orthogroup Method | OrthoFinder | Generates one-to-one, one-to-many, and many-to-many orthologs | Method choice affects orthogroup completeness |
The following diagram illustrates the complete OMG workflow from single-cell data processing to cross-species cell type prediction:
Table: Essential Research Resources for Implementing OMG Analysis
| Resource Type | Specific Tool/Resource | Function in OMG Analysis |
|---|---|---|
| Computational Framework | R Statistical Environment | Primary platform for data analysis and visualization |
| Single-Cell Analysis | Seurat R Package | Cell clustering and marker gene identification |
| Orthology Prediction | OrthoFinder | Generation of orthologous gene groups across species |
| Precomputed Orthogroups | 15-Species Orthogroup Set | Reference orthogroups for common plant species |
| Implementation Code | OMG R Package on GitHub | Custom functions for OMG conversion and statistical testing |
| Web Accessibility Tool | OMG Browser | User-friendly web interface for cell type identification |
Comparative Analysis: OMG Versus Alternative Approaches
Performance Evaluation Across Plant Lineages
The OMG method has been rigorously validated against traditional approaches using well-annotated single-cell maps from Arabidopsis, tomato, rice, and maize [5]. The following table summarizes the quantitative performance comparisons:
Table: Performance Comparison of Cell Type Identification Methods
| Method | Basis of Comparison | Arabidopsis-Tomato(Dicot-Dicot) | Arabidopsis-Rice(Dicot-Monocot) | ComputationalEfficiency | Handling of GeneFamily Expansions |
|---|---|---|---|---|---|
| OMG Approach | Orthologous Marker Groups | 12/13 clusters correct(92% accuracy) | 13/14 cluster pairs fromorthologous cell types | High (no integration needed) | Excellent (explicitly designed for this) |
| One-to-One OrthologIntegration | Sequence Similarity Only | Limited data | 3/8 cluster pairs fromorthologous cell types | Moderate | Poor (fails with duplicated genes) |
| SAMap Method | Gene-Gene Bipartite Graph | Limited data | Limited data | Low (requires iterative refinement) | Moderate |
| Co-expression Proxies | Expression Pattern Similarity | Limited data | Limited data | Moderate to Low | Moderate |
Experimental validation between Arabidopsis and tomato roots demonstrated that the OMG method successfully recapitulated manually annotated cell types with high accuracy [5]. In the dicot-dicot comparison, all 13 tomato root clusters showed exact, partial, or functional matches with corresponding Arabidopsis clusters [5]. Particularly noteworthy was the method's ability to identify functional similarities between distinct cell types—tomato exodermis clusters showed significant OMG overlap with Arabidopsis endodermis, both of which contain suberized barriers in their cell walls [5].
In the more evolutionarily challenging dicot-monocot comparison between Arabidopsis and rice, the OMG method identified 14 pairs of cell clusters with significant similarities, with 13 of these pairs representing orthologous cell types [5]. This performance substantially exceeded that of one-to-one ortholog mapping, which only identified 8 cluster pairs with just 3 representing orthologous cell types [5].
Large-Scale Validation Across Diverse Plant Species
The robustness of the OMG method was further demonstrated in a comprehensive analysis mapping 1 million cells across 268 cell clusters from 15 diverse plant species [5] [30]. This large-scale application revealed 14 dominant groups with substantial conservation in shared cell-type markers across both monocots and dicots [5]. The success of this expansive comparison highlights the method's scalability and utility for studying evolutionary conservation of cell types across deep phylogenetic distances.
The method's statistical framework effectively distinguished true biological conservation from random matches, with four mismatched clusters in the Arabidopsis-rice comparison subsequently identified as meristematic cells through additional validation [5]. Gene Ontology enrichment analysis confirmed these clusters shared functional characteristics, including enrichment for ribosomal genes characteristic of meristematic identity [5].
Experimental Protocols and Implementation Guidelines
Detailed Methodological Protocol
For researchers implementing the OMG method, the following step-by-step protocol provides a reproducible framework:
Data Preprocessing and Marker Gene Identification
- Process single-cell RNA-seq data using standard pipelines (e.g., Seurat)
- Perform clustering using established algorithms
- Identify top 200 marker genes for each cluster using appropriate statistical tests
Orthologous Group Construction
- For species not included in precomputed sets: gather protein sequences and run OrthoFinder (
orthofinder -f OrthoFinder_protein_folder) - Load orthogroups file (
Orthogroups.tsv) into R environment - Clean orthogroups for each species using provided functions (
clean_OG())
- For species not included in precomputed sets: gather protein sequences and run OrthoFinder (
Orthologous Marker Group Conversion
- Merge marker genes with orthogroups using
merge_MG_OG()function - This converts species-specific marker genes into evolutionarily conserved OMGs
- Merge marker genes with orthogroups using
Cross-Species Comparison
- Perform pairwise comparisons between species using
test_significant()function - Set appropriate FDR threshold (typically 0.01-0.05)
- Generate visualization plots using
generate_plot_comparison()
- Perform pairwise comparisons between species using
Cell Type Prediction and Validation
- Extract significant OMG overlaps using
extract_table() - Predict cell identities based on conserved orthologous markers
- Validate predictions using known markers or experimental approaches
- Extract significant OMG overlaps using
Practical Implementation Considerations
The selection of N=200 marker genes per cluster represents a carefully optimized balance between specificity and sensitivity [5]. This parameter ensures sufficient overlapping OMGs between species while preserving marker gene specificity. The method's implementation is available through both command-line R packages and a user-friendly web-based OMG browser, making it accessible to researchers with varying computational expertise [5] [30].
For studies focusing on specific biological processes, the OMG framework can be supplemented with functional enrichment analysis using tools like Gene Ontology enrichment to validate the biological relevance of identified conserved cell types [5]. This integrated approach strengthens conclusions about functional conservation beyond mere transcriptional similarity.
The Orthologous Marker Group method represents a significant advancement in comparative plant transcriptomics, addressing fundamental challenges posed by gene family expansions and divergent expression patterns across species. By leveraging orthogroup conservation rather than simple sequence similarity, OMG analysis enables robust cell type identification across evolutionarily divergent species where traditional methods fail.
The method's strong performance across dicot-dicot and dicot-monocot comparisons, coupled with its successful application to a 15-species atlas encompassing 1 million cells, demonstrates its broad utility for evolutionary developmental biology studies [5]. The availability of user-friendly implementations, including the OMG browser, ensures this approach will be accessible to the broader plant research community [5].
Future developments will likely expand the reference database to include additional species, incorporate multi-omics data layers, and enhance statistical frameworks for identifying more subtle forms of conservation. As single-cell technologies continue to advance and be applied to non-model organisms, approaches like OMGs will play an increasingly vital role in unraveling the evolution of cell types and developmental programs across the plant kingdom.
The dramatic reduction in sequencing costs has led to an explosion of genomic and transcriptomic data for non-model plant species, creating unprecedented opportunities for comparative evolutionary studies [31]. However, downstream bioinformatic analyses remain a significant bottleneck, requiring specialized expertise and substantial computational resources that are inaccessible to many researchers [31] [32]. Plant genome annotations vary considerably in quality due to differences in assembly methods, genome complexity, and available resources, further complicating cross-species comparisons [31]. Within this context, orthogroup conservation analysis has emerged as a fundamental approach for understanding gene family evolution, gene function diversification, and genome duplication events across plant lineages [31] [33].
Table: Comparative Analysis of Plant Gene Family Resources
| Resource | Primary Function | Data Input Limitations | Key Strengths | Plant-Specific Optimizations |
|---|---|---|---|---|
| PlantTribes2 | Gene family classification & evolutionary analysis | Scalable for genome-wide datasets | Modular workflow, Galaxy integration, duplication inference | Yes, with plant-specific gene family scaffolds |
| PLAZA 5.0 | Comparative genomics platform | Max 300 sequences for family assignment | Curated plant genomes, functional annotations | Yes, focused on plant species |
| OrthoMCL-DB | Ortholog group database | Limited to pre-computed species | Broad taxonomic coverage, established method | Limited plant-specific adjustments |
| PlantTribes (v1) | Gene family database | Static classification | Historical plant gene families, phylogenetic trees | Yes, but outdated |
Several computational frameworks have been developed to address these challenges, yet most suffer from critical limitations in scalability, accessibility, or plant-specific optimization [31]. The original PlantTribes database, developed in 2008, provided a foundational resource with gene family classifications for five sequenced plant species but became outdated as sequencing technologies advanced [34] [35]. Static databases like PLAZA 5.0 offer rich functional annotations but severely restrict user data input—allowing only 300 external sequences for gene family assignment—making them unsuitable for analyzing new genomes or transcriptomes [31] [32]. This landscape creates a pressing need for flexible, scalable tools that can handle the growing volume of plant genomic data while providing robust evolutionary context.
PlantTribes2 Framework: Architecture and Innovations
PlantTribes2 represents a comprehensive reimplementation of the original PlantTribes concept, transforming it from a static relational database into a flexible analytical pipeline with entirely new codebase, features, and extensive testing [31]. At its core, PlantTribes2 utilizes objectively classified gene family scaffolds derived from high-quality plant genomes, which serve as reference clusters for sorting and analyzing new sequences [31] [32]. The framework accepts multiple entry points, including assembled transcriptomes, gene model predictions, or individual coding sequences, making it adaptable to various research scenarios and data types [31].
A key innovation in PlantTribes2 is its modular workflow architecture, which enables researchers to perform comprehensive gene family analyses through interconnected analytical steps [31]. The pipeline begins with optional transcript model improvement, then proceeds to orthologous group assignment using pre-computed gene family clusters with rich functional annotation information [32]. For gene families of interest, PlantTribes2 subsequently enables multiple sequence alignment, gene family phylogeny reconstruction, estimation of synonymous and non-synonymous substitution rates, and inference of large-scale duplication events [31] [32]. This complete workflow provides an integrated solution for evolutionary genomics investigations in plants.
Table: PlantTribes2 Analytical Modules and Outputs
| Analysis Module | Primary Algorithms/Tools | Key Outputs | Evolutionary Applications |
|---|---|---|---|
| Gene Family Assignment | OrthoMCL, BLAST-based sorting | Orthogroup classification, functional annotations | Orthology determination, gene copy number variation |
| Multiple Sequence Alignment | MUSCLE, MAFFT | Protein/DNA alignments | Conserved motif identification, evolutionary rates |
| Gene Family Phylogeny | Maximum likelihood methods | Phylogenetic trees with support values | Duplication event inference, phylogenetic context |
| Evolutionary Rates | Codon-based models | dN/dS ratios, selective pressure | Positive selection detection, functional divergence |
| Duplication Inference | Syntery analysis, tree reconciliation | Gene duplication events, ages | Genome evolution, neofunctionalization |
Accessibility represents another major advancement in PlantTribes2. The developers have collaborated with the Galaxy Project to create wrappers for all PlantTribes2 tools, making them available through the main public Galaxy instance (usegalaxy.org) and deployable in any Galaxy instance [31] [32]. This web-based framework eliminates the command-line barrier for many researchers while maintaining computational power through connection to high-performance computing resources [31]. Additionally, PlantTribes2 is available for download via GitHub and Bioconda, providing multiple access points for researchers with different computational preferences and resources [31] [36].
Comparative Performance Analysis
Scalability and Data Input Handling
When evaluated against other gene family analysis resources, PlantTribes2 demonstrates superior scalability characteristics, particularly for analyzing new genomic datasets. Unlike static databases with strict input limitations, PlantTribes2 can process genome-scale datasets without arbitrary restrictions on sequence numbers [31]. This capability was demonstrated in a case study on Rosaceae genomes, where researchers successfully classified gene families across 15 genomes from 6 genera, creating a valuable community resource that was previously unavailable [33]. The scalable architecture enables researchers to work with increasingly large datasets as sequencing technologies continue to advance.
In contrast, popular databases like PLAZA 5.0, while containing carefully selected high-quality plant genomes with rich functional annotations, impose significant constraints on user data input [31] [32]. The platform restricts users to uploading a maximum of 300 new sequences for BLAST-based gene family searches and adding only 50 external sequences when constructing gene family phylogenies on their web server [31] [32]. These limitations make genome-scale analyses of new datasets impractical with such static resources. PlantTribes2 effectively bridges this gap by providing both curated reference data and flexible analytical capacity for user-provided sequences.
Workflow Integration and Analytical Comprehensive
PlantTribes2 provides a comprehensive analytical ecosystem that surpasses the capabilities of specialized tools with narrower foci. The framework integrates multiple analytical steps into a cohesive workflow, from initial sequence quality improvement through sophisticated evolutionary analyses [31]. This end-to-end integration contrasts with approaches that require researchers to manually combine disparate tools, often with incompatible outputs or significant technical barriers [31]. The unified workflow reduces analytical overhead and ensures consistency across different stages of gene family investigation.
The Core Orthogroup (CROG) analysis capability of PlantTribes2 enables particularly powerful investigations of conserved gene families across multiple species [31] [33]. This approach was effectively demonstrated in a study of economically important Rosaceae species, where researchers identified conserved orthogroups related to architectural traits while simultaneously revealing significant issues with existing genome assemblies [33]. By implementing an iterative curation workflow, the researchers corrected thousands of missing genes resulting from methodological biases in the 'Bartlett' pear genome, dramatically improving gene family representations for downstream functional studies [33].
Experimental Applications and Case Studies
Transcriptomic Analysis in Orobanchaceae
PlantTribes2 has been successfully applied to transcriptomic studies in the weedy Orobanchaceae family, demonstrating its utility for analyzing complex non-model organisms [31]. In this application, researchers utilized the pipeline to classify assembled transcript sequences into orthologous gene families, enabling evolutionary comparisons across species with diverse ecological adaptations [31]. The scalable framework efficiently handled the transcriptomic data, sorting sequences into pre-computed orthologous groups while leveraging rich functional annotation information from reference genomes [31]. This approach facilitated the identification of lineage-specific gene family expansions potentially associated with parasitic lifestyles in these species.
The Orobanchaceae case study highlighted PlantTribes2's capability to work with assembled transcriptomes rather than requiring complete genome sequences, significantly broadening its applicability to non-model species where only transcriptomic data are available [31]. The pipeline's ability to improve transcript models before orthogroup assignment further enhanced the quality of subsequent evolutionary analyses [31]. This application demonstrates how PlantTribes2 enables robust comparative genomic investigations even in taxonomically challenging groups with limited genomic resources.
Rosaceae Genome Analysis and Gene Model Curation
In a comprehensive study of Rosaceae genomes, researchers employed PlantTribes2 to build a foundation for gene family analysis across 15 genomes from 6 genera [33]. This work exemplified the pipeline's utility for cross-genome comparisons and identification of orthologs and paralogs in an economically important plant family [33]. The researchers developed a novel workflow incorporating PlantTribes2 for targeted improvement of gene models related to tree architecture in European pear genomes, laying the groundwork for future functional studies [33].
A particularly significant outcome of this research was the discovery and correction of widespread annotation issues in existing reference genomes [33]. The comparative gene family approach revealed that the most recent 'Bartlett' pear genome assembly contained thousands of missing genes due to methodological bias [33]. After implementing global-scale assembly corrections, the researchers used PlantTribes2 for targeted improvement of architecture-related genes in both 'Bartlett' and 'd'Anjou' pear genomes [33]. This case demonstrates how integrating comparative genomics and phylogenomics through PlantTribes2 can dramatically enhance gene annotation quality and facilitate gene discovery in important crop species.
Experimental Protocols and Methodologies
Core Orthogroup Analysis Workflow
The standard workflow for orthogroup conservation analysis using PlantTribes2 involves sequential stages that can be implemented through the Galaxy interface or command-line tools. The initial stage consists of data preparation and quality control, where protein coding sequences from the species of interest are compiled and filtered for completeness [31] [33]. For transcriptomic data, this stage may include transcript model improvement using external evidence such as protein homology or expression support [31].
The core analytical stage begins with gene family assignment, where sequences are sorted into pre-computed orthologous gene family clusters using a combination of similarity searches and graph-based clustering algorithms [31] [34]. The resulting orthogroups then undergo multiple sequence alignment using tools like MUSCLE or MAFFT to establish positional homology for subsequent analyses [31] [34]. For phylogenetic inference, PlantTribes2 employs maximum likelihood methods to reconstruct gene family trees, which serve as the basis for inferring duplication events and evolutionary relationships [31] [34]. Additional modules estimate evolutionary rates (dN/dS ratios) to identify signatures of selective pressure and perform large-scale duplication inference through synteny analysis or tree reconciliation approaches [31].
Gene Model Curation Protocol
For researchers working with newly sequenced or poorly annotated genomes, PlantTribes2 enables a rigorous gene model curation protocol that was successfully applied in Rosaceae genomes [33]. The process begins with genome assembly and annotation using standard tools, followed by comparative gene family classification across multiple related species using PlantTribes2 [33]. Researchers then identify anomalies in gene family sizes or distributions that may indicate missing or erroneous gene models in the target species [33].
The curation phase involves targeted gene model improvement using a combination of transcriptomic evidence, protein homology, and conserved synteny information [33]. Corrected gene models are then re-integrated into the PlantTribes2 classification to verify their appropriate placement within orthogroups [33]. This iterative process continues until gene family representations are biologically plausible based on phylogenetic relationships and known genome duplication histories [33]. The final output consists of curated gene families with accurate gene models suitable for functional and evolutionary inferences [33].
Research Reagent Solutions
Table: Essential Research Reagents and Resources for PlantTribes2 Analysis
| Resource Type | Specific Examples | Function in Analysis | Availability |
|---|---|---|---|
| Reference Genomes | Arabidopsis thaliana, Oryza sativa, Pyrus communis | Provide scaffold for gene family classification | PLAZA, NCBI, Phytozome |
| Sequence Alignment Tools | MUSCLE, MAFFT | Generate multiple sequence alignments | Integrated in PlantTribes2 |
| Phylogenetic Inference | Maximum likelihood implementations | Reconstruct gene family trees | Integrated in PlantTribes2 |
| Orthology Methods | OrthoMCL, graph clustering | Define orthologous groups | Integrated in PlantTribes2 |
| Computational Environment | Galaxy Workbench, Bioconda | Execution environment for analyses | usegalaxy.org, GitHub |
PlantTribes2 represents a significant advancement in plant comparative genomics, addressing critical limitations in scalability, accessibility, and analytical comprehensive faced by existing resources [31]. Its modular workflow architecture enables end-to-end gene family analysis from sequence data to evolutionary inferences, while integration with the Galaxy platform dramatically reduces technical barriers for biological researchers [31] [32]. The framework's demonstrated applications in diverse contexts—from transcriptomics in Orobanchaceae to genome-wide orthogroup analysis in Rosaceae—highlight its versatility for addressing various evolutionary questions in plant biology [31] [33].
As plant genomics continues to expand into increasingly diverse species with complex genomes, tools like PlantTribes2 will play an essential role in extracting evolutionary insights from genomic data [31] [33]. The scalable design ensures compatibility with growing dataset sizes, while the open availability through multiple distribution channels promotes widespread adoption and community development [31] [36]. For researchers investigating orthogroup conservation across plant species, PlantTribes2 provides a comprehensive, accessible solution that bridges the gap between rapidly accumulating sequence data and meaningful biological understanding.
In the field of comparative plant genomics, accurately inferring evolutionary relationships between genes across multiple species is fundamental to understanding trait diversity, adaptation mechanisms, and evolutionary history. Orthogroup analysis addresses this need by identifying sets of genes descended from a single ancestral gene in the last common ancestor of the species being considered [10]. This approach provides a critical framework for comparative genomics, enabling researchers to trace gene lineage evolution across speciose plant families, identify conserved gene networks, and discover lineage-specific genetic innovations [37] [5]. For plant researchers investigating phenomena such as stress response conservation, developmental pathway evolution, or metabolic diversification, robust orthogroup inference forms the essential foundation for meaningful comparative analysis.
The computational workflow from raw sequence data to orthogroup assignment presents significant challenges, including gene length biases, differential evolutionary rates, gene duplication events, and incomplete lineage sorting [13] [10]. This guide objectively compares the performance of leading orthogroup inference methods, with particular emphasis on OrthoFinder's capabilities within plant research contexts. We provide experimental data from independent benchmarks and detailed methodologies to assist researchers in selecting and implementing appropriate workflows for their specific plant genomic studies.
Orthogroup Inference Methods: Capabilities and Performance Comparison
Various computational methods have been developed to infer orthogroups from genomic data, each employing distinct algorithms to address the challenges of homology inference. OrthoMCL utilizes BLAST for sequence similarity searches followed by the Markov Cluster algorithm to identify orthogroups, though it exhibits significant gene length bias that affects accuracy [10]. OMA employs a pairwise approach focused on identifying orthologs between species pairs, providing high precision but potentially lower recall for complete orthogroup discovery [13] [10]. InParanoid specializes in identifying orthologs and in-paralogs between two species, making it suitable for focused comparisons but less ideal for multi-species analyses [13]. OrthoFinder implements a phylogenetically-driven approach that identifies orthogroups and subsequently infers gene trees, rooted species trees, and gene duplication events [13]. A key innovation in OrthoFinder is its normalization procedure that eliminates gene length bias in orthogroup detection, significantly improving accuracy over methods like OrthoMCL [10].
Performance Benchmarking on Standardized Datasets
Independent benchmarking studies provide critical insights into the relative performance of orthogroup inference methods. The Orthobench database, comprising 70 expert-curated reference orthogroups (RefOGs) across bilaterian species, serves as a gold standard for evaluation [38]. When applied to this benchmark, OrthoFinder demonstrated substantially improved accuracy compared to other methods:
Table 1: Orthogroup Inference Accuracy on Orthobench Benchmark
| Method | Precision | Recall | F-score | Gene Length Bias |
|---|---|---|---|---|
| OrthoFinder | Substantially increased across all sequence lengths | Dramatically improved for short sequences | 8-33% higher than other methods | Eliminated through score normalization |
| OrthoMCL | High for medium-length sequences | Low for short sequences | Moderate to low | Strong bias observed |
| OMA | High precision | Low recall for complete orthogroups | Moderate | Not reported |
| InParanoid | High for pairwise comparisons | Limited to two-species analysis | Context-dependent | Not reported |
Recent re-evaluation of the Orthobench dataset using improved phylogenetic methods revealed that 44% of the original RefOGs required revision (31 of 70), with 24 needing major changes and 7 requiring minor modifications [38]. This highlights both the challenge of orthogroup inference and the importance of using updated benchmarks and methods that leverage improved phylogenetic approaches.
Beyond orthogroup inference, OrthoFinder excels at ortholog identification. In the Quest for Orthologs benchmark, OrthoFinder achieved 3-24% higher accuracy on the SwissTree test and 2-30% higher accuracy on the TreeFam-A test compared to other methods [13]. This performance advantage stems from its phylogenetic approach, which better distinguishes variable sequence evolution rates from true phylogenetic relationships.
Workflow Implementation: From Sequences to Orthogroups
Comprehensive OrthoFinder Workflow
The OrthoFinder workflow transforms raw protein sequences into comprehensively annotated orthogroups through a multi-stage process. The following diagram illustrates the complete analytical pipeline:
Figure 1: OrthoFinder workflow from sequence input to comprehensive orthology inference
Detailed Workflow Stages
Orthogroup Inference: OrthoFinder begins by performing all-vs-all sequence similarity searches using DIAMOND or BLAST, then applies a novel score normalization that eliminates gene length bias [10]. The normalized scores are used to build a graph of sequence similarities which is clustered to identify orthogroups.
Gene Tree Inference: For each orthogroup, multiple sequence alignments are generated and used to infer phylogenetic trees. While OrthoFinder uses DendroBLAST by default for speed, it supports integration with other alignment and tree inference tools like MAFFT and IQ-TREE for maximum accuracy [13].
Species Tree Inference: OrthoFinder automatically infers a rooted species tree from the complete set of gene trees using a novel algorithm that does not require prior knowledge of species relationships [13].
Gene Tree Rooting and Analysis: The rooted species tree enables accurate rooting of all gene trees, which is essential for proper interpretation of orthology and paralogy relationships. The rooted trees are analyzed using a duplication-loss-coalescence (DLC) model to identify orthologs and gene duplication events [13].
This comprehensive workflow produces not only orthogroups but also gene trees, the rooted species tree, gene duplication events, and extensive comparative genomics statistics, providing plant researchers with a complete phylogenetic framework for their analyses [13].
Experimental Protocols and Benchmarking Methodologies
Standardized Benchmarking Protocols
To ensure fair and informative comparisons between orthogroup inference methods, the scientific community has developed standardized benchmarking approaches:
Orthobench Protocol:
- Dataset Curation: 70 expert-curated reference orthogroups (RefOGs) spanning the Bilateria with diverse challenges for inference methods [38].
- Sequence Collection: Latest proteomes for the original 12 species plus additional outgroup and ingroup species for improved phylogenetic context.
- Sequence Search: HMMER searches using HMMs from the original study with liberal e-value thresholds to maximize inclusion of potential orthologs.
- Phylogenetic Analysis: Multiple sequence alignment using MAFFT L-INS-i algorithm, alignment trimming with TrimAL, and tree inference with IQ-TREE using best-fit substitution models [38].
- Orthogroup Delineation: Manual curation and phylogenetic assessment to determine true Bilateria-level orthogroup membership.
Quest for Orthologs Benchmarking:
- Reference Dataset: 2011_04 dataset with results from multiple methods for comprehensive comparison [13].
- Accuracy Assessment: Precision, recall, and F-score calculations against gold-standard trees in SwissTree and TreeFam-A tests.
- Discordance Evaluation: Species Tree Discordance Tests (STDT and GSTDT) assessing percentage of trials where orthologs are identified and Robinson-Foulds distance between species trees and gene trees [13].
Implementation in Plant Research Contexts
For plant-specific applications, researchers have adapted these protocols:
Phylotranscriptomic Analysis:
- Orthogroup Identification: OrthoFinder analysis of protein sequences from multiple plant species to identify orthogroups [37].
- Expression Analysis: Integration of RNA-seq data to identify conserved cold-responsive transcription factor orthogroups (CoCoFos) across eudicots [37].
- Functional Validation: Experimental validation through expression analysis and genetic manipulation, as demonstrated for BBX29 as a negative regulator of cold tolerance in Arabidopsis [37].
Single-Cell Transcriptomics Conservation:
- Marker Identification: Top N marker genes (typically 200) identified for each cell cluster in single-cell RNA-seq data [5].
- Orthologous Marker Groups: OrthoFinder used to generate orthologous gene groups across multiple plant species [5].
- Conservation Analysis: Statistical testing (Fisher's exact test) to identify significant overlaps in orthologous marker groups between cell clusters across species, revealing conserved cell identity programs [5].
Comparative Performance Data
Quantitative Accuracy Assessment
Independent benchmarking provides critical performance data for orthogroup inference methods:
Table 2: Comprehensive Performance Comparison of Orthogroup Inference Methods
| Method | Orthobench Precision | Orthobench Recall | SwissTree Accuracy | TreeFam-A Accuracy | Species Tree Accuracy | Computational Speed |
|---|---|---|---|---|---|---|
| OrthoFinder | High (length-independent) | High (length-independent) | 3-24% higher than competitors | 2-30% higher than competitors | Automated and accurate | Equivalent to fastest score-based methods |
| OrthoMCL | Variable (length-dependent) | Low for short genes | Moderate | Moderate | Not provided | Fast |
| OMA | High | Low for complete orthogroups | Moderate | Moderate | Not provided | Moderate |
| Hieranoid | Not reported | Not reported | Moderate | Moderate | Not provided | Fast |
| SonicParanoid | Not reported | Not reported | Moderate | Moderate | Not provided | Very fast |
Specialized Applications in Plant Research
OrthoFinder has enabled numerous advances in plant comparative genomics:
Cross-Species Cell Identity Conservation: Researchers developing Orthologous Marker Gene Groups (OMGs) for cell type identification across plant single-cell transcriptomes utilized OrthoFinder to generate orthologous groups across 15 diverse plant species [5]. This approach successfully identified 14 dominant groups with substantial conservation in shared cell-type markers across monocots and dicots, demonstrating the utility of OrthoFinder for complex evolutionary analyses in plants.
Transcription Factor Classification: OrthoFinder has been used to provide a complete classification of transcription factor gene families in plants, revealing 6.9 million previously unobserved relationships [10]. This comprehensive analysis provides plant researchers with an invaluable resource for understanding the evolution of transcriptional regulation across plant species.
Woody Plant Genomics: The Woody Plant Multi-Omics Database (WP-MOD) integrates OrthoFinder analysis for 373 species across 35 orders, providing gene ortholog identification as a core feature for comparative analysis of woody species [39]. This implementation highlights how OrthoFinder scales to accommodate the large genomic datasets characteristic of plant comparative genomics.
Successful implementation of orthogroup analysis requires specific computational tools and resources:
Table 3: Essential Research Reagent Solutions for Orthogroup Analysis
| Resource | Function | Application Context | Implementation Notes |
|---|---|---|---|
| OrthoFinder | Phylogenetic orthology inference | Core orthogroup identification across species | Default uses DIAMOND for speed; supports multiple aligners and tree inference tools [13] |
| DIAMOND | Accelerated sequence similarity search | Alternative to BLAST for large datasets | Significantly faster than BLAST with comparable sensitivity [13] |
| MAFFT | Multiple sequence alignment | Gene tree construction | L-INS-i algorithm recommended for accuracy [38] |
| IQ-TREE | Phylogenetic tree inference | Gene tree and species tree construction | ModelFinder selects best-fit substitution models [38] |
| Orthobench | Benchmarking dataset | Method validation and comparison | 70 curated orthogroups; recently updated with phylogenetic revisions [38] |
| WP-MOD | Woody plant multi-omics database | Plant-specific genomic context | Integrates genomes, orthogroups, and analysis tools for 373 woody species [39] |
| OMG Browser | Orthologous marker group analysis | Cell type conservation across species | Web-based tool for plant single-cell transcriptomics [5] |
Orthogroup inference represents a fundamental analytical step in plant comparative genomics, with significant implications for understanding gene family evolution, conserved regulatory networks, and species-specific adaptations. Through objective benchmarking and performance comparison, OrthoFinder demonstrates superior accuracy in both orthogroup and ortholog inference, while providing a comprehensive phylogenetic framework that extends beyond simple clustering approaches.
The implementation of robust workflows from sequence data to orthogroup assignment, as detailed in this guide, empowers plant researchers to conduct more accurate and biologically informative comparative analyses. As genomic datasets continue to grow in both size and taxonomic breadth, methods like OrthoFinder that combine computational efficiency with phylogenetic rigor will become increasingly essential for extracting meaningful biological insights from complex genomic data.
For researchers embarking on plant orthogroup analyses, we recommend: (1) utilizing the most current genome annotations available; (2) implementing OrthoFinder with appropriate computational resources for large-scale plant genomes; (3) validating results using conserved gene families with established phylogenetic relationships; and (4) leveraging plant-specific resources like WP-MOD for taxonomic context and functional annotation. Through careful implementation of these workflows and resources, plant researchers can reliably uncover the evolutionary patterns and processes that have shaped the genomic diversity of the plant kingdom.
In plant single-cell RNA sequencing (scRNA-seq) studies, a fundamental challenge is the accurate identification of cell types, which is considerably more complex than in animal systems. This difficulty arises from the scarcity of known cell-type marker genes and the fact that even the closest orthologs of established markers from model species like Arabidopsis often do not conserve their cell-type-specific expression patterns across diverse plant lineages [40] [5]. Furthermore, gene family expansions through frequent tandem duplication and whole-genome duplication events in plants complicate the use of standard one-to-one ortholog mapping approaches that work successfully in mammalian systems [40] [5].
Traditional computational approaches for cross-species cell type identification have significant limitations in plant applications. Integration-based methods that project data from multiple species into a shared manifold space require substantial computational resources and can produce clusters with mixed cell identities when applied to diverse plant species [40]. These methods also necessitate careful selection of reference species and cannot identify cell types that don't exist in the chosen reference [40]. To address these challenges, a novel computational strategy called Orthologous Marker Gene Groups (OMGs) was developed specifically for plant systems, enabling robust cell-type identification and comparison across diverse species without requiring cross-species data integration [40] [5].
Methodological Framework: The OMG Approach
Core Workflow and Implementation
The OMG method employs a systematic, three-stage workflow for cross-species cell type identification that leverages orthologous gene groups rather than individual orthologs [40] [5]:
Marker Gene Identification: The top N marker genes (typically N=200) are identified for each cell cluster in each species using established tools like Seurat [40] [5]. Empirical validation has demonstrated that using 200 markers provides an optimal balance between sufficient overlapping markers across diverse species and preservation of marker gene specificity [40].
Orthologous Group Generation: OrthoFinder is employed to generate orthologous gene groups across multiple plant species (15 species in the initial implementation), encompassing one-to-one, one-to-many, and many-to-many orthologous relationships that are particularly important in plants due to frequent gene duplication events [40] [5].
Statistical Significance Testing: Pairwise comparisons are performed using overlapping OMGs between each cluster in the query species and reference species, with Fisher's exact test and false discovery rate (FDR) correction used to identify clusters with statistically significant numbers of shared OMGs [40] [5].
The following diagram illustrates the complete OMG analytical workflow:
The OMG Browser: Accessibility for Practicing Biologists
To make the OMG method accessible to the broader plant research community, the developers created a user-friendly web-based tool called the OMG Browser [40] [41]. This platform provides a no-code workflow that allows researchers to upload cluster marker genes, select appropriate reference species and tissues, and obtain intuitive heatmap visualizations showing statistical significance for each cluster-to-cell-type comparison [41]. The browser implementation significantly lowers the barrier for plant biologists to apply sophisticated orthologous marker analysis without requiring specialized computational expertise [41].
Experimental Application and Validation
Performance Benchmarking Against Alternative Methods
The OMG method was rigorously validated through multiple experiments comparing its performance against traditional one-to-one ortholog mapping and integration-based approaches [40]. In a critical test comparing cell type identification between Arabidopsis (dicot) and rice (monocot) root scRNA-seq datasets, the OMG method demonstrated superior performance [40].
Table 1: Performance Comparison Between OMG and One-to-One Ortholog Methods for Arabidopsis-Rice Root Cell Type Identification
| Method | Significant Cluster Pairs Identified | Orthologous Cell Type Pairs | Exact or Partial Matches | Key Limitations |
|---|---|---|---|---|
| OMG Method | 14 pairs | 13 out of 14 (93%) | 9 pairs | Limited discrimination for undifferentiated cell clusters |
| One-to-One Ortholog Mapping | 8 pairs | 3 out of 8 (38%) | Not reported | High rate of erroneous annotations |
When applied to dicot-dicot comparisons (Arabidopsis and tomato roots), the OMG method identified 24 pairs of clusters with significant numbers of shared OMGs (FDR < 0.01) [40]. The resulting annotations showed exact matches for 12 tomato clusters, one partial match (cortex cluster shared significant OMGs with both cortex and nonhair cells in Arabidopsis), and functional matches for two exodermis clusters (a cell type not found in Arabidopsis) that shared significant OMGs with endodermis clusters in Arabidopsis [40]. This functional relationship makes biological sense as both cell types contain suberized barriers in their cell walls [40].
Large-Scale Application Across 15 Plant Species
The most comprehensive validation of the OMG method involved its application to an unprecedented dataset comprising over 1 million cells organized into 268 cell clusters across 15 diverse plant species [40]. This large-scale analysis revealed 14 dominant groups with substantial conservation in shared cell-type markers across both monocots and dicots [40] [5]. The successful application at this scale demonstrates the robustness and scalability of the OMG approach for comparative plant biology at phylogenetic depths not previously achievable.
Table 2: Summary of OMG Method Validation Across Diverse Experimental Setups
| Validation Scenario | Species Compared | Clusters Analyzed | Annotation Accuracy | Key Findings |
|---|---|---|---|---|
| Dicot-Dicot Comparison | Arabidopsis vs. Tomato | 15 × 11 = 165 pairs | Exact or functional matches for all clusters | Exodermis (tomato) showed functional match with endodermis (Arabidopsis) |
| Monocot-Dicot Comparison | Arabidopsis vs. Rice | 11 × 10 clusters | 13/14 orthologous cell types | Superior to one-to-one ortholog mapping |
| Pan-Species Analysis | 15 plant species | 268 clusters | 14 conserved cell-type groups | Broad conservation across monocots and dicots |
Comparative Analysis with Alternative Approaches
The OMG method occupies a distinct niche in the landscape of computational approaches for cross-species cell type identification. Unlike integration-based methods such as SAMap [40] or approaches that identify co-expression proxies [40], the OMG method does not require computationally intensive construction and iteration of gene-gene bipartite graphs [40]. This fundamental difference makes the OMG approach particularly suitable for plant species with expanded gene families and frequent genome duplication events.
Table 3: Method Comparison for Cross-Species Cell Type Identification in Plants
| Method | Computational Requirements | Handling of Gene Family Expansions | Required Expertise | Best Application Context |
|---|---|---|---|---|
| OMG Method | Lower (no integration) | Excellent (uses orthogroups) | Low (web browser available) | Broad cross-species comparisons |
| Integration-Based Methods (SAMap, etc.) | Higher (manifold alignment) | Problematic (relies on orthologs) | High (computational expertise) | Closely related species |
| One-to-One Ortholog Mapping | Low | Poor (uses single orthologs) | Medium | Very closely related species |
| Literature-Derived Marker Sets | Low | Variable (depends on curation) | Low | Species with well-annotated markers |
Essential Research Tools and Reagents
Successful application of the OMG method relies on several key computational tools and resources that form the core infrastructure for orthologous marker analysis.
Table 4: Essential Research Reagent Solutions for OMG Analysis
| Tool/Resource | Function | Key Features | Implementation in OMG |
|---|---|---|---|
| OrthoFinder | Orthogroup inference | Handles one-to-one, one-to-many, many-to-many orthologs | Generates orthologous gene groups across species |
| Seurat | scRNA-seq analysis and marker detection | Cell clustering, differential expression | Identifies top marker genes for each cell cluster |
| OMG Browser | User-friendly analysis interface | No-code workflow, heatmap visualization | Accessible cell type annotation for non-specialists |
| R Package (GitHub) | Custom analysis implementation | Parameter adjustment, extended functionality | Flexible implementation for experienced users |
Integration with Broader Transcriptomics Technologies
The OMG method complements recent advances in plant single-cell transcriptomics, including the development of single-nucleus RNA sequencing (snRNA-seq) as an alternative to protoplast-based scRNA-seq [42] [43]. While scRNA-seq analyzes entire cells, providing a complete picture of gene expression including cytoplasmic RNA, snRNA-seq focuses solely on nuclear RNA, eliminating the need for protoplast preparation and avoiding potential stress responses triggered by cell isolation methods [42] [43]. Both approaches can feed into the OMG pipeline for cross-species comparison.
Similarly, emerging spatial transcriptomics technologies like Stereo-seq (with resolution of 500 nm) are revealing gene expression patterns within the native tissue context [42]. When combined with single-cell transcriptomics, these spatial methods enable mapping of gene expression from individual cells to their precise tissue locations [44]. The OMG method can leverage the cell-type markers validated through these spatial approaches to enhance the accuracy of orthologous cell type identification.
The OMG method represents a significant advancement in comparative plant cell biology by enabling reliable cell-type identification across deeply diverged plant species. Its unique approach of using orthologous marker groups rather than individual orthologs effectively addresses the challenges posed by frequent gene duplication events in plant genomes. The method's scalability has been demonstrated through its successful application to over 1 million cells across 15 species, revealing 14 conserved cell-type groups across monocots and dicots.
The development of the user-friendly OMG Browser makes this sophisticated analytical approach accessible to plant biologists without computational expertise, potentially transforming how the community approaches cell-type annotation in non-model species. As single-cell and spatial transcriptomics technologies continue to advance, generating increasingly comprehensive atlases of plant cellular diversity, methods like OMG will play a crucial role in deciphering the conservation and divergence of cell types across the plant kingdom. This comparative framework provides a powerful foundation for understanding the evolutionary mechanisms that generate cellular diversity in plants and how these relate to adaptation and diversification.
Optimizing Orthogroup Analysis: Addressing Technical Challenges and Parameter Selection
In plant species research, the identification of orthologous genes—those related by vertical descent from a common ancestor—has long been a cornerstone of comparative genomics and functional annotation. Traditionally, this process has heavily relied on single-copy orthologs, where gene families contain exactly one representative sequence per species. This conservative approach aims to minimize the confounding effects of paralogs (genes related through duplication events) on phylogenetic inference and functional prediction. However, this methodological constraint severely limits the amount of data available for analysis, particularly as the number of species in a study increases. Recent advances in genomic sequencing and computational methods now enable researchers to move beyond this limitation by incorporating data from expanded gene families, unlocking greater phylogenetic resolution and functional insights. This guide compares the performance of traditional single-copy ortholog approaches with emerging methods that leverage complete gene family data, providing plant researchers with experimental data and protocols to inform their orthogroup conservation analyses.
Quantitative Comparison of Orthology Inference Approaches
The table below summarizes the key differences between the three primary approaches for orthology inference in phylogenetic studies, based on recent comparative research.
Table 1: Performance Comparison of Orthology Inference Methods
| Methodological Approach | Data Utilization | Computational Demand | Paralog Handling | Best Application Context |
|---|---|---|---|---|
| Single-Copy Orthologs (SCOs) | Limited (typically 5-15% of genes) | Low | Exclusion only | Conservative studies with closely-related species |
| Tree-Based Decomposition | Moderate to high (30-50% of genes) | High | Explicit identification and extraction | High-resolution phylogenies with quality genomes |
| All Gene Families (orthologs + paralogs) | Maximum (near 100% of genes) | Moderate | Robust inclusion with specialized methods | Phylogenomic analyses with duplication-rich histories |
The data clearly demonstrates that using all gene families drastically increases the number of genes available for phylogenetic analysis compared to single-copy approaches. One study comparing primate genomes found that while single-copy families included only 1,820 genes, analyzing all gene families provided access to over 428,129 gene copies—an increase of more than two orders of magnitude [45]. Similar patterns have been observed in plant studies, where the expansion of gene families has been linked to important adaptive traits [46].
Experimental Protocols for Comprehensive Orthogroup Analysis
Protocol 1: Orthogroup Inference and Filtering
This protocol outlines the standard workflow for identifying orthogroups across multiple plant species, based on methodologies successfully applied in large-scale comparative genomic studies.
Table 2: Essential Research Reagents and Tools for Orthogroup Analysis
| Research Tool | Specific Function | Application Notes |
|---|---|---|
| OrthoFinder | Identifies orthogroups from protein sequences | Default parameters typically sufficient; supports multi-threading |
| BUSCO | Assesses genome completeness | Use lineage-specific datasets for plants (e.g., embryophyta_odb10) |
| MAFFT | Generates multiple sequence alignments | Preferred for accuracy with divergent sequences |
| IQ-TREE | Infers phylogenetic trees | ModelFinder extension automatically selects best substitution model |
| TRIMAL | Trims ambiguous regions from alignments | Prevents overestimating evolutionary distances |
Step-by-Step Workflow:
- Data Preparation: Obtain protein sequences for all species in FASTA format. Use tools like
primary_transcript.py(included with OrthoFinder) to retain only the longest transcript per gene. - Quality Assessment: Evaluate genome completeness using BUSCO with plant-specific lineage datasets. Only include genomes with >80% completeness for reliable orthogroup inference.
- Orthogroup Identification: Run OrthoFinder with default parameters. The algorithm performs an all-vs-all BLAST search, clusters sequences into orthogroups using the MCL algorithm, and infers a rooted species tree.
- Orthogroup Filtering: Exclude orthogroups present in fewer than 10% of taxa to minimize species-specific artifacts [47].
- Downstream Analysis: Export orthogroup sequences for phylogenetic analysis, calculate gene family expansion/contraction rates using CAFE, or perform functional enrichment analysis.
This protocol was successfully applied in a study of 1,154 yeast genomes, where 5,551 core orthogroups were identified containing approximately 90% of all genes assigned to orthogroups [47]. In plant studies, similar approaches have revealed how gene family expansions regulate context-dependent symbiotic interactions [46].
Protocol 2: Phylogenetic Inference with Multi-Copy Gene Families
This protocol specifically addresses the challenge of incorporating multi-copy gene families into phylogenetic analyses.
Experimental Workflow:
- Gene Tree Construction: For each orthogroup, align protein sequences using MAFFT, trim ambiguous regions with TRIMAL, and infer gene trees using IQ-TREE with automatic model selection.
- Orthology Extraction (Optional): For tree-based decomposition approaches, use tools like LOFT or DISCO to identify duplication nodes and extract orthologous subtrees [45].
- Species Tree Inference: Use methods robust to paralogs such as ASTRAL (a quartet-based method) for analyzing multi-copy gene families. Alternatively, use concatenation approaches with site-heterogeneous models.
- Support Assessment: Calculate branch support using multi-locus bootstrapping or local posterior probabilities depending on the species tree method used.
Studies have demonstrated that quartet-based methods like ASTRAL remain robust even when paralogs are included, because the most common quartet is still expected to match the species tree despite gene duplication and loss events [45]. This approach has been validated across diverse datasets including primates, fungi, and plants [45].
Visualization of Methodological Workflows
The following diagram illustrates the key decision points and methodological options when designing a study that moves beyond single-copy orthology limitations:
Case Studies in Plant Genomics
Gene Family Expansions in Plant-Microbe Interactions
Research on arbuscular mycorrhizal (AM) symbiosis across 42 angiosperms demonstrated that gene family expansions provide molecular flexibility for context-dependent regulation of species interactions [46]. Plants that associate with mycorrhizal fungi showed expanded gene families with up to 200% more context-dependent gene expression and double the genetic variation associated with mycorrhizal benefits to plant fitness. Notably, these expansions arose primarily from tandem duplications rather than whole-genome duplications, creating a continuous source of genetic variation that allows fine-tuning of symbiotic interactions throughout plant evolution [46].
Biosynthetic Gene Clusters in Medicinal Plants
Comparative genomics of medicinal plants has revealed that biosynthetic gene clusters (BGCs)—physical groupings of genes involved in specialized metabolic pathways—often arise through gene family expansions [48]. For instance, the biosynthesis of complex therapeutic compounds like taxol, vinblastine, and artemisinin involves expanded and co-localized gene families that have evolved through duplication and neofunctionalization events [48]. These expansions enable plants to produce structurally diverse molecules that are largely beyond the reach of chemical synthesis, highlighting the importance of understanding gene family evolution for drug discovery and development [49].
The limitations of single-copy orthology approaches are increasingly apparent as plant genomics advances. Methods that incorporate data from all gene families—whether through tree-based decomposition or paralog-robust species tree inference—provide access to substantially more phylogenetic information while maintaining analytical accuracy. Quantitative comparisons demonstrate that these approaches can increase usable data by 10-20 times compared to single-copy methods. For plant researchers conducting orthogroup conservation analyses, embracing these expanded methodologies will be essential for uncovering the genetic basis of adaptive traits, specialized metabolism, and evolutionary innovations that have shaped the plant kingdom.
Orthologous Marker Gene Groups (OMGs) represent a novel computational strategy for identifying cell types in both model and non-model plant species, enabling rapid comparison across published single-cell maps without requiring cross-species data integration [5]. This method addresses a fundamental challenge in plant single-cell RNA sequencing (scRNA-seq): the scarcity of known cell-type marker genes and the divergence of marker expression patterns across species, which severely limits accurate cell-type identification and investigations into cellular conservation [5] [50]. The OMG framework specifically tackles the limitations of existing approaches, including the unreliability of one-to-one orthologous genes for data integration due to gene family expansions and duplications common in plants, and the significant computational demands of integration-based methods like SAMap [5].
At its core, the OMG method operates on the principle that while individual marker genes may not conserve their expression patterns across divergent species, the collective signal from groups of orthologous genes (orthogroups) retains sufficient information to accurately determine cell identity [5]. This approach leverages the observation that functional similarity between orthologous genes is often higher than between paralogs, a concept known as the ortholog conjecture, which has been supported by evidence showing high conservation of tissue-specificity between orthologs across species [51]. By shifting the unit of comparison from individual genes to orthologous groups, the OMG method achieves a more robust balance between specificity and sensitivity—two critical parameters that often exist in a trade-off relationship in ortholog prediction methods [52].
Orthologous Marker Groups: Methodological Framework and Workflow
Core Computational Protocol
The OMG method follows a structured computational workflow that transforms raw single-cell data into annotated cell types through systematic comparative analysis:
Cluster Marker Identification: For each cell cluster in the query species, the top N marker genes (typically N=200) are identified using established approaches such as Seurat [5] [50]. This uniform number ensures statistical comparability between cell clusters.
Orthologous Group Construction: OrthoFinder is employed to generate orthologous gene groups across M plant species (M=15 in the initial study) [5]. This step accommodates one-to-one, one-to-many, and many-to-many orthologous relationships, crucial for addressing gene family expansions common in plants.
Statistical Significance Testing: Pairwise comparisons are performed using overlapping OMGs between each cluster in the query species and reference species. Results are visualized via heatmaps showing -log10(FDR) values from Fisher's exact tests, which determine clusters with significant numbers of shared OMGs [5] [41].
The selection of N=200 marker genes represents a critical balancing point in the OMG workflow. Empirical testing revealed that using fewer than 200 markers leads to a rapid decrease in overlapping OMGs across diverse species, reducing sensitivity, while using more than 200 markers diminishes specificity [5]. This parameter optimization ensures sufficient overlapping markers while preserving marker gene specificity across evolutionary distances.
Workflow Visualization
Performance Benchmarking: OMG Versus Alternative Approaches
Comparative Analysis of Ortholog Prediction Methods
The OMG method was rigorously validated against established approaches using well-annotated single-cell maps from Arabidopsis, tomato, rice, maize, and poplar [5]. The benchmarking focused on key performance metrics including annotation accuracy, ability to handle evolutionary divergence, and computational efficiency.
Table 1: Performance comparison of cell type identification methods across species
| Method | Core Approach | Annotation Accuracy (Tomato vs Arabidopsis) | Annotation Accuracy (Rice vs Arabidopsis) | Handles Gene Family Expansions | Computational Demand |
|---|---|---|---|---|---|
| OMG Method | Orthologous Marker Groups | 12/12 clusters exact/partial match [5] | 13/14 cluster pairs orthologous types [5] | Excellent (incorporates one-to-many, many-to-many) [5] | Low (no integration required) [5] |
| One-to-One Ortholog Integration | Single ortholog mapping | Not reported | 3/8 pairs orthologous types [5] | Poor (fails with duplication events) [5] | Moderate |
| SAMap | Gene-gene bipartite graph | Not reported | Not reported | Moderate | High (iterative refinement) [5] |
| Co-expression Proxies | Co-expression proxy pairs | Not reported | Not reported | Moderate | High [5] |
When comparing dicot species (Arabidopsis and tomato), the OMG method successfully identified 24 pairs of clusters with significant shared OMGs, with published annotations of 12 tomato clusters exactly matching corresponding Arabidopsis clusters [5]. The cortex cluster in tomato showed partial match, sharing significant OMGs with both cortex and nonhair cells in Arabidopsis, while two exodermis clusters (a cell type not found in Arabidopsis) showed significant OMG overlap with endodermis clusters—a functionally meaningful connection as both cell types contain suberized barriers in their cell walls [5].
In more evolutionarily distant comparisons between dicots and monocots (Arabidopsis and rice), the advantage of OMGs over one-to-one ortholog mapping became particularly pronounced. While one-to-one orthologous genes identified significant similarities between only 8 pairs of cell clusters with limited accuracy, the OMG method identified 14 pairs with significant similarities, 13 of which represented orthologous cell types [5]. The four mismatched clusters were located in connecting regions of UMAPs, suggesting they contained undifferentiated cells, a hypothesis supported by subsequent GO enrichment analysis showing ribosomal gene enrichment—a hallmark of meristematic identity [5].
Specificity and Sensitivity Quantification
The OMG method demonstrates an exceptional balance between specificity and sensitivity, achieving high annotation accuracy across 15 diverse plant species encompassing 1 million cells and 268 cell clusters [5]. The method revealed 14 dominant groups with substantial conservation in shared cell-type markers across monocots and dicots, demonstrating its ability to identify evolutionarily conserved cellular programs while maintaining specificity sufficient to distinguish closely related cell types [5].
This performance advantage stems from the method's fundamental design: by using orthologous groups rather than individual genes as the unit of comparison, OMG inherently buffers against evolutionary divergence in individual marker genes while amplifying the conserved signal through group-wise analysis. This approach aligns with the established principle that orthologs tend to have higher functional similarity than paralogs, particularly in terms of tissue-specificity, which evolves slowly in the absence of duplication events [51].
Table 2: Specificity and sensitivity indicators in ortholog prediction methods
| Method | Sensitivity Indicator | Specificity Indicator | Overall Performance |
|---|---|---|---|
| OMG Method | Identified 14 conserved cell groups across 15 species [5] | 13/14 correct orthologous pairs in Arabidopsis-rice comparison [5] | Excellent balance, enables cross-species annotation [5] |
| InParanoid | Lower average proteome size in human-mouse analysis [52] | High conservation of gene order [52] | Good specificity, lower sensitivity [52] |
| OrthoMCL | Moderate average proteome size [52] | High conservation of gene order [52] | Good balance [52] |
| KOG Best Pair | Lowest average proteome size [52] | Highest expression correlation [52] | High specificity, low sensitivity [52] |
| PhyloGenetic Tree (PGT) | Large average proteome size [52] | Low conservation of gene order [52] | High sensitivity, lower specificity [52] |
Essential Research Reagents and Computational Tools
Successful implementation of OMG analysis requires specific computational tools and resources that enable the various stages of the workflow, from initial single-cell processing to final orthologous group comparison.
Table 3: Essential research reagents and computational tools for OMG analysis
| Tool/Resource | Function | Key Features | Application in OMG Workflow |
|---|---|---|---|
| Seurat | Single-cell analysis | Cell clustering, marker gene identification [5] | Identifies top N marker genes per cell cluster [5] |
| OrthoFinder | Orthogroup inference | Phylogenetic orthogroup inference [5] | Generates orthologous gene groups across species [5] |
| OMG Browser | Web-based annotation | User-friendly interface, no coding required [41] | Upload marker genes, view significance heatmaps [41] |
| Fisher's Exact Test | Statistical testing | Determines significance of OMG overlap [5] | Quantifies cluster-to-cell-type similarity [5] |
| Single-Cell Reference Atlas | Reference data | Curated single-cell maps across species [5] | Provides reference for cross-species comparison [5] |
The OMG Browser deserves particular attention as it provides a user-friendly web interface that implements the complete OMG workflow without requiring programming expertise [41]. Researchers can upload cluster marker genes, select appropriate reference species and tissues, and receive a heatmap visualization showing statistical significance for each cluster-to-cell-type comparison, with significant matches clearly highlighted for biological interpretation [41].
Experimental Design and Implementation Guidelines
Optimized Experimental Protocol
Based on the validation studies conducted in the original OMG research, the following protocol represents an optimized framework for implementing OMG analysis:
Single-Cell Data Preprocessing and Clustering
- Process raw single-cell RNA-seq data using standard preprocessing steps (quality control, normalization, dimensionality reduction)
- Perform cell clustering using graph-based methods (e.g., Seurat) at biologically reasonable resolution
- Identify top 200 marker genes for each cluster using conservative thresholds (minimum percent expressed, effect size) [5]
Orthologous Group Construction
- Select appropriate reference species (15 species were used in original study) [5]
- Run OrthoFinder on protein sequences from all query and reference species
- Validate orthogroup assignments through manual inspection of key marker genes
Cross-Species Comparison and Annotation
- Select appropriate reference single-cell atlas based on tissue and phylogenetic proximity
- Calculate overlapping OMGs between query clusters and reference cell types
- Perform Fisher's exact test for each cluster-type pair with multiple testing correction
- Annotate clusters based on significant matches (FDR < 0.01)
- Validate annotations using orthogonal evidence when possible (in situ hybridization, known developmental trajectories) [41]
Critical Parameter Optimization
The OMG method's performance depends heavily on several key parameters that require careful optimization:
Marker Gene Selection (N=200) The choice of 200 marker genes per cluster represents a carefully optimized balance. Experimental validation showed that values below 200 rapidly decrease overlapping markers across species (reduced sensitivity), while values above 200 diminish specificity [5]. Researchers should maintain this parameter unless working with exceptionally high- or low-quality datasets.
Statistical Significance Threshold The use of Fisher's exact test with FDR correction at q < 0.01 provides stringent control of false discoveries while maintaining power to detect evolutionarily conserved relationships [5]. This threshold successfully eliminated spurious matches in validation studies while retaining biologically meaningful connections.
Clustering Resolution Extremely fine clustering can produce small clusters with too few reliable markers, reducing annotation confidence [41]. Initial analysis should use biologically reasonable clustering resolution, with refinement based on marker gene robustness and biological plausibility of resulting annotations.
The OMG method represents a significant advancement in plant single-cell genomics by providing a robust framework for cross-species cell type annotation that effectively balances specificity and sensitivity. By shifting the unit of comparison from individual genes to orthologous groups, the method overcomes fundamental limitations of previous approaches, particularly their inability to handle frequent gene family expansions and duplications in plant genomes.
The demonstrated performance across 15 diverse plant species, encompassing monocots and dicots, highlights the method's broad applicability for evolutionary and developmental studies [5]. The identification of 14 dominant cell groups with substantial conservation suggests deep evolutionary preservation of core cellular identities across angiosperms, opening new avenues for investigating the conservation and divergence of developmental programs.
The availability of a user-friendly web interface (OMG Browser) makes this powerful approach accessible to plant biologists without computational expertise, potentially accelerating single-cell research in non-model species [41]. As single-cell transcriptomics expands to encompass more plant diversity, the OMG framework provides a essential tool for unlocking the comparative potential of these data, enabling researchers to move beyond descriptive cataloging toward mechanistic insights into the evolution of plant form and function.
Future methodological developments will likely focus on integrating additional data types beyond transcriptomics, incorporating spatial information, and extending the orthologous group concept to regulatory elements. As the field progresses, the balance between specificity and sensitivity that OMGs provide will remain a central consideration in the development of increasingly powerful comparative cellular genomics methods.
In the field of plant comparative genomics, the accurate identification of conserved orthogroups—sets of genes descended from a single gene in a last common ancestor—is fundamental to understanding evolutionary relationships and functional conservation across species [10]. This process inherently involves testing thousands of gene clusters for significant conservation, creating a substantial multiple testing challenge that requires robust statistical validation. Without proper correction, a considerable proportion of statistically significant findings are likely to be false positives, potentially leading to erroneous biological conclusions [53] [54].
This guide provides an objective comparison of statistical validation methodologies centered on the implementation of Fisher's Exact Test combined with False Discovery Rate (FDR) correction, a widely adopted approach in genomics research [5]. We present experimental data and practical protocols to help researchers select appropriate statistical frameworks for orthogroup conservation analysis, ensuring both rigorous validation and biologically meaningful discovery.
Statistical Framework Comparison
Core Statistical Concepts
Table 1: Key Statistical Methods for Multiple Hypothesis Testing
| Method | Control Type | Primary Application | Advantages | Limitations |
|---|---|---|---|---|
| False Discovery Rate (FDR) | Expected proportion of false discoveries among rejected hypotheses [53] | Genome-wide studies (e.g., orthogroup conservation) [5] | Greater power than FWER methods; allows some false positives [53] [54] | Less stringent than FWER; requires careful interpretation [53] |
| Family-Wise Error Rate (FWER) | Probability of at least one false discovery [53] | Clinical trials; safety-critical applications | Strong control over any false positives [53] | Overly conservative for genomic studies; low power [53] [54] |
| Bonferroni Correction | FWER control through significance threshold adjustment [53] | Small number of hypothesis tests | Simple implementation; strong error control [53] | Dramatically reduces power with thousands of tests [53] |
| Benjamini-Hochberg Procedure | FDR control through step-up p-value adjustment [54] | High-throughput genomics; transcriptomics [54] | Balance between discovery and false positives; widely adopted [53] [54] | Requires independent or positively correlated tests [54] |
Quantitative Performance Metrics
Table 2: Statistical Power Comparison Across Multiple Testing Corrections
| Correction Method | Theoretical Basis | True Positive Rate* | False Positive Control* | Suitable Number of Tests |
|---|---|---|---|---|
| Uncorrected | Raw p-values (α = 0.05) | High | Very Poor | Not recommended for multiple tests |
| Bonferroni | Single-step adjustment [53] | Very Low | Excellent | < 100 |
| Holm-Bonferroni | Step-down adjustment [54] | Low | Excellent | 100-1,000 |
| Benjamini-Hochberg (FDR) | Sequential p-value rejection [54] | Moderate-High | Good | > 1,000 (ideal for genomics) |
| Benjamini-Yekutieli | FDR under arbitrary dependence [54] | Moderate | Good | > 1,000 with unknown dependence |
*Relative performance indicators based on typical genomic studies with thousands of simultaneous tests [53]
Experimental Protocols for Orthogroup Conservation Analysis
Workflow for Statistical Validation of Orthogroups
The following diagram illustrates the complete experimental workflow for orthogroup conservation analysis with integrated statistical validation:
Implementation of Fisher's Exact Test
Fisher's Exact Test is particularly valuable for orthogroup conservation analysis as it tests the null hypothesis that the overlap between two gene sets occurs by random chance, making it ideal for determining significant conservation between orthogroups across species [5].
Protocol: Fisher's Exact Test for Orthogroup Overlap
Construct Contingency Table: For each pair of orthogroups from different species, create a 2×2 contingency table:
- Cell A: Genes in both orthogroups (overlap)
- Cell B: Genes in orthogroup 1 but not orthogroup 2
- Cell C: Genes in orthogroup 2 but not orthogroup 1
- Cell D: Genes in neither orthogroup (background)
Calculate Hypergeometric Probability: Compute the exact probability of observing the given distribution under the null hypothesis of no association:
(P = \frac{(A+B)!(C+D)!(A+C)!(B+D)!}{A!B!C!D!N!})
Where N represents the total number of genes in the analysis.
Interpret Results: A significant p-value (typically after FDR correction) indicates non-random overlap suggesting evolutionary conservation [5].
FDR Control Implementation
The Benjamini-Hochberg procedure provides a straightforward method for FDR control in orthogroup analyses:
Protocol: Benjamini-Hochberg FDR Control
Order P-Values: Sort all raw p-values from m hypothesis tests in ascending order: (P{(1)} \leq P{(2)} \leq \ldots \leq P_{(m)})
Apply Sequential Threshold: For a chosen FDR level α (typically 0.05), find the largest k such that: (P_{(k)} \leq \frac{k}{m} \alpha)
Reject Hypotheses: Reject all null hypotheses (H{(1)}, \ldots, H{(k)}) corresponding to (P{(1)}, \ldots, P{(k)}) [54]
This procedure ensures that the expected proportion of false discoveries among all significant findings is at most α [53] [54].
Comparative Experimental Data
Case Study: Cross-Species Orthogroup Analysis
Table 3: Performance Comparison in Plant Orthogroup Identification [5]
| Analysis Method | Statistical Approach | Significant Cluster Pairs Identified | Validation Rate* | Computational Efficiency |
|---|---|---|---|---|
| One-to-One Orthologs | Fisher's Exact Test + FDR < 0.01 | 8 pairs | 37.5% | High |
| Orthologous Marker Groups (OMGs) | Fisher's Exact Test + FDR < 0.01 | 14 pairs | 92.9% | Moderate |
| Direct Data Integration | Unspecified significance threshold | 24 regions with mixed identities | 58.3% | Low |
| OrthoFinder + OMG | Fisher's Exact Test + FDR < 0.01 [5] | 13 orthologous cell types | 92.3% | Moderate-High |
*Validation rate indicates percentage of identified pairs with confirmed orthologous relationships through manual curation or functional analysis [5]
Research Reagent Solutions
Table 4: Essential Tools for Orthogroup Analysis and Statistical Validation
| Tool/Resource | Function | Application Context | Key Features |
|---|---|---|---|
| OrthoFinder [10] | Orthogroup inference | Identifying homologous genes across species | Solves gene length bias; dramatically improves accuracy |
| Plant Duplicate Gene Database [55] | Data resource | Plant-specific orthogroup analysis | Classifies duplication modes across 141 plant genomes |
| Custom R/Python Scripts | Statistical implementation | FDR control and Fisher's Exact Test | Flexible implementation of Benjamini-Hochberg procedure |
| Orthologous Marker Groups (OMGs) [5] | Cell type identification | Cross-species cell type comparison | Enables comparison without data integration |
| OrthoBench [10] | Benchmark dataset | Method validation | Manually curated orthogroups for performance assessment |
Technical Considerations and Best Practices
Statistical Implementation Guidelines
The relationship between statistical tests, multiple testing correction, and biological interpretation can be visualized as follows:
Application Condition Considerations
FDR control is specifically appropriate when:
- Conducting hundreds or thousands of hypothesis tests simultaneously [53] [56]
- Seeking to balance discovery of true positives with control of false positives [54]
- Performing exploratory research where follow-up validation is planned [53]
Alternative approaches (e.g., FWER control) may be preferable when:
- Testing a small number of pre-specified hypotheses [56]
- The consequences of any false positive are severe [53]
- Conducting confirmatory research where no follow-up validation is planned [56]
The integration of Fisher's Exact Test with FDR correction provides a statistically robust yet biologically sensitive framework for orthogroup conservation analysis in plant genomics. This approach enables researchers to identify evolutionarily conserved gene sets with controlled false discovery rates, balancing statistical rigor with discovery power. As plant genomic datasets continue to expand in both scale and complexity, these statistical validation methods will remain essential for distinguishing meaningful biological conservation from random noise in comparative genomic studies.
Computational Resource Management for Large-Scale Cross-Species Comparisons
Large-scale cross-species genomic comparisons represent a cornerstone of modern plant evolutionary biology and functional genomics. These analyses enable researchers to trace the conservation and diversification of gene families, regulatory elements, and genome architecture across evolutionary timescales. The computational management of such comparisons presents significant challenges in terms of data heterogeneity, scaling across phylogenetic distances, and method selection. This guide objectively evaluates available computational resources and methods specifically designed for orthogroup conservation analysis across plant species, providing performance comparisons and detailed experimental protocols to assist researchers in selecting appropriate tools for their specific research contexts. As genomic datasets continue to expand in both scale and taxonomic breadth, efficient computational resource management becomes increasingly critical for generating biologically meaningful insights from cross-species comparative analyses.
Computational Method Landscape for Cross-Species Plant Genomics
The field of cross-species plant genomics has developed diverse computational approaches to address the specific challenges posed by plant genome architecture, including frequent whole-genome duplications, tandem gene expansions, and varied genome sizes. These methods can be broadly categorized into sequence-based, expression-based, and structure-based approaches, each with distinct resource requirements and applications in orthogroup conservation analysis.
Table 1: Computational Methods for Cross-Species Plant Genomics
| Method Category | Representative Tools | Primary Application | Key Strengths |
|---|---|---|---|
| Genome Skimming | varKoder, Skmer, iDeLUCS | Taxonomic identification & phylogenetic placement | Handles low-coverage sequencing; applicable to degraded DNA [57] |
| Ortholog Identification | OrthoFinder, OMG Method | Orthogroup inference & cell type identification | Handles complex gene families; accounts for gene duplications [40] |
| 3D Genome Analysis | 3D-GDP Database | Conserved regulatory structure identification | Comparative analysis of TADs, loops, and compartments [58] |
| Single-Cell Integration | SAMap, Co-expression Proxies | Cross-species cell atlas comparison | Maps cell types across divergent species [40] |
Among these approaches, orthogroup inference represents a particularly critical resource for cross-species comparisons, with OrthoFinder having emerged as a standard tool for identifying orthologous relationships across multiple species [40]. The recently developed Orthologous Marker Gene Groups (OMG) method extends this approach specifically for single-cell RNA sequencing analysis, enabling cell type identification and comparison across diverse plant species without requiring resource-intensive cross-species data integration [40].
Performance Benchmarking and Experimental Data
Benchmarking Datasets for Method Evaluation
Standardized benchmarking datasets are essential for objective performance comparisons of computational methods. The plant genomics community has developed several curated resources specifically for this purpose:
The varKoder benchmark dataset provides a standardized resource for evaluating genome skimming tools, comprising four distinct datasets with varying phylogenetic depths [57]. This includes newly sequenced Malpighiales data (287 accessions, 195 species), species-level datasets for plants (Corallorhiza orchids), animals (Bembidion beetles), fungi, and bacteria (Mycobacterium tuberculosis), as well as eukaryotic family-level data from NCBI SRA [57]. This hierarchical dataset structure enables researchers to test method performance across different taxonomic scales from closely related populations to distantly related families.
For orthogroup inference, OrthoBench has served as the standard benchmarking dataset for orthogroup inference algorithms for over a decade [57]. This resource supports the development and evaluation of computational methods for interpreting genetic variants and functional sequences, with particular relevance for plant species with complex gene family expansions.
Performance Metrics and Comparative Data
The OMG method for cross-species cell type identification has demonstrated robust performance across diverse plant lineages. When applied to single-cell data from Arabidopsis and tomato roots, the method identified 24 pairs of clusters with significant numbers of shared OMGs (FDR < 0.01), with published annotations of 12 clusters in tomato exactly matching corresponding Arabidopsis clusters [40]. The method showed particular strength in identifying functionally related cell types even when exact orthology was unclear, such as connecting exodermis clusters in tomato with endodermis clusters in Arabidopsis based on shared suberized barriers in cell walls [40].
Table 2: Performance Metrics for Cross-Species Computational Methods
| Method | Accuracy Metrics | Dataset Scope | Computational Requirements |
|---|---|---|---|
| OMG Method | 13/14 correct cluster pairs between Arabidopsis and rice; 9 exact/partial matches [40] | 15 species, 268 cell clusters, 1 million cells [40] | Low (no integration required) |
| varKoder | Effective from closely-related (0.6 My) to distantly-related (34.1 My) species [57] | 195 Malpighiales species; all taxa in NCBI SRA [57] | Medium (handles low-coverage genomes) |
| OrthoFinder | Benchmarking via OrthoBench [57] | Flexible (M=15 species in OMG implementation) [40] | Medium to High (scales with species number) |
The OMG method demonstrated significant advantages over one-to-one ortholog approaches, particularly when comparing distantly related species. In tests between Arabidopsis (dicot) and rice (monocot), one-to-one orthologous genes identified significant similarities between only 8 pairs of cell clusters, with just 3 from orthologous cell types [40]. In contrast, the OMG method identified 14 cluster pairs with significant similarities, with 13 from orthologous cell types and 9 representing exact or partial matches [40]. This performance advantage stems from the method's ability to handle complex orthologous relationships including one-to-many and many-to-many correspondences that are common in plant genomes.
Experimental Protocols for Cross-Species Comparisons
OMG Method Workflow for Cell Type Conservation
The Orthologous Marker Gene Groups (OMG) method provides a robust protocol for identifying conserved cell types across plant species without computational intensive data integration. The following workflow details the implementation for orthogroup conservation analysis:
Step 1: Marker Gene Identification
- Process single-cell RNA sequencing data using standard tools (e.g., Seurat)
- Identify top N marker genes for each cell cluster (N=200 recommended)
- Recommendation of N=200 balances sufficient overlapping OMGs with marker gene specificity [40]
Step 2: Orthologous Group Generation
- Employ OrthoFinder to generate orthologous gene groups for M plant species
- Include both closely and distantly related species in the orthogroup analysis
- The OMG implementation used M=15 species with high-quality single-cell maps [40]
Step 3: Statistical Testing for Conservation
- Perform pairwise comparisons using overlapping OMGs between clusters
- Apply Fisher's exact test with FDR correction for multiple comparisons
- Visualize results using heatmaps showing -log10(FDR) values
- Clusters with FDR < 0.01 considered statistically significant [40]
This protocol successfully identified 14 dominant groups with substantial conservation in shared cell-type markers across monocots and dicots, demonstrating the power of orthogroup-based approaches for cross-species comparisons in plants [40].
Figure 1: OMG Method Workflow for Cross-Species Cell Type Identification
Genome Skimming Protocol for Taxonomic Identification
For molecular identification across species using low-coverage genome sequencing, the following protocol based on varKoder development provides a standardized approach:
Step 1: Dataset Curation
- Select taxa representing appropriate phylogenetic depth (closely-related to distantly-related)
- Include taxonomically verified samples for validation
- The Malpighiales dataset includes 287 accessions representing 195 species [57]
Step 2: Sequence Processing
- Obtain raw genome skim sequences from public repositories (NCBI SRA) or new sequencing
- Generate two-dimensional graphical representations of genomic data
- Alternative approaches include chaos game representations or k-mer frequency profiles
Step 3: Method Application and Validation
- Apply multiple identification methods to the same dataset (e.g., varKoder, Skmer, iDeLUCS)
- Compare to conventional DNA barcodes assembled with tools like PhyloHerb
- Validate using taxonomically verified samples and calculate accuracy metrics
This protocol has been successfully applied to datasets spanning from closely related populations (e.g., Stigmaphyllon species with 0.6-34.1 Myr divergence) to all eukaryotic families in NCBI SRA [57].
Research Reagent Solutions
The following table details essential computational reagents and resources for implementing large-scale cross-species comparisons in plant genomics:
Table 3: Essential Research Reagents for Cross-Species Genomic Comparisons
| Resource Type | Specific Resource | Function in Research | Access Information |
|---|---|---|---|
| Reference Dataset | varKoder Benchmark Dataset | Standardized data for method validation & comparison | Malpighiales data: 287 accessions, 195 species [57] |
| Software Tool | OrthoFinder | Orthogroup inference across multiple species | Standard for orthogroup identification [40] |
| Database | 3D-GDP Database | Comparative analysis of 3D genome structures | http://www.3d-gdp.com/ [58] |
| Analysis Method | OMG R Package | Cell type identification & conservation analysis | GitHub repository [40] |
| Web Tool | OMG Browser | User-friendly cell type identification | Web-based interface for biologists [40] |
These resources collectively provide a comprehensive toolkit for researchers conducting cross-species comparisons in plants. The benchmark datasets enable method validation and comparison, while specialized databases like 3D-GDP offer insights into conserved three-dimensional genome architectures across 26 plant species [58]. The combination of command-line tools and user-friendly web interfaces ensures accessibility for researchers with varying computational expertise.
Effective computational resource management for large-scale cross-species comparisons in plant genomics requires careful selection of methods matched to specific biological questions and dataset characteristics. Orthogroup-based approaches like the OMG method provide significant advantages for identifying conserved cellular programs across evolutionary distances, while genome skimming methods enable taxonomic placement across diverse phylogenetic scales. The development of standardized benchmarking datasets and specialized plant genomics databases continues to enhance the reproducibility and robustness of cross-species comparative genomics. As these methods evolve, integration across genomic scales - from sequence conservation to three-dimensional genome architecture - will provide increasingly comprehensive insights into the evolutionary principles shaping plant diversity.
In the field of plant genomics, the accuracy and completeness of genome assemblies fundamentally shape the validity of downstream biological insights, especially in comparative studies such as orthogroup conservation analysis. High-quality reference genomes are essential for correctly identifying orthologous genes and gene families across species, which in turn illuminates evolutionary relationships, conserved cellular functions, and the genetic basis of traits [5] [59]. This guide objectively compares the primary methods and metrics used for genome quality assessment, focusing on the interplay between BUSCO completeness and other assembly metrics, and their collective impact on cross-species genomic research.
Core Concepts in Genome Assessment
Genome assembly quality is evaluated across three primary dimensions, often termed the "3 Cs": Contiguity, Completeness, and Correctness [60].
- Contiguity describes the physical connectedness of the assembly, measured by metrics like contig N50 (the length of the shortest contig at 50% of the total genome length). Higher N50 values indicate a more connected, less fragmented assembly [60].
- Completeness assesses what proportion of the expected genome is present. BUSCO (Benchmarking Universal Single-Copy Orthologs) is a standard tool that evaluates this by searching for a set of evolutionarily conserved, single-copy genes that should be present in nearly all members of a specific taxonomic lineage [61] [59].
- Correctness refers to the accuracy of the assembled sequence at the base-pair level. This includes the absence of misassemblies, insertions, deletions, and base errors [60].
These dimensions are not independent; a comprehensive assessment requires integrating all three to understand the assembly's true utility for research.
Table 1: Key research reagents and computational tools for genome assembly and assessment.
| Tool/Resource Name | Primary Function | Role in Genome Analysis |
|---|---|---|
| BUSCO [61] [59] | Completeness Assessment | Quantifies the presence of universal single-copy orthologs to estimate gene space completeness. |
| OrthoFinder [5] | Orthogroup Inference | Identifies orthologous groups of genes across multiple species, fundamental for comparative genomics. |
| metaSPAdes [62] | Genome Assembly | Performs de novo assembly of genome sequences from metagenomic or standard sequencing data. |
| Merqury & Yak [60] | Correctness Assessment | Uses k-mer comparisons between assembly and short reads to count errors and assess base-level accuracy. |
| OrthoDB [61] | Ortholog Database | Provides the curated sets of orthologous genes that BUSCO uses as a benchmark. |
BUSCO and Assembly Metrics in Practice
Interpreting BUSCO Results
BUSCO classifies genes into four categories, providing a clear, interpretable summary of genome completeness [61]:
- Complete (Single-Copy): The ideal finding, indicating the core gene is present and complete in a single copy.
- Complete (Duplicated): The gene is found complete, but in multiple copies. This can indicate true biological duplication, assembly artifacts, or unresolved heterozygosity [61] [60].
- Fragmented: Only a portion of the BUSCO gene was found, suggesting the assembly is interrupted or the gene sequence is of low quality in that region.
- Missing: The BUSCO gene is entirely absent, pointing to a significant gap in the assembly [61].
A high percentage of complete BUSCOs is the primary indicator of a high-quality assembly. However, a high duplication rate can signal issues like contamination or over-assembly, while many fragmented or missing genes suggest the assembly is incomplete or suffers from low sequence continuity [61].
Comparative Analysis of Assessment Metrics
While BUSCO is excellent for assessing gene space completeness, it must be used alongside other metrics to get a full picture of assembly quality. The following table synthesizes how different metrics work together.
Table 2: A comparison of key metrics for genome assembly assessment.
| Metric | What It Measures | Strengths | Limitations | Ideal Outcome |
|---|---|---|---|---|
| BUSCO Completeness [61] [59] | Presence of conserved core genes. | Standardized, biologically meaningful, taxon-aware. | Only assesses a small, conserved subset of the genome. | >95% complete, single-copy BUSCOs. |
| Contig N50 [60] | Physical continuity and fragmentation of the assembly. | Intuitive measure of assembly scaffold length. | Does not reflect base-level accuracy or completeness. | As high as possible (e.g., >1 Mb). |
| Transcript Mappability [59] | Accuracy and completeness of gene models. | Directly assesses the quality of the functionally important gene space. | Requires high-quality RNA-seq data; only assesses genic regions. | High alignment rate, coverage, and depth. |
| K-mer Based Correctness (e.g., Merqury) [60] | Base-level accuracy by comparing with short-read data. | Assesses the entire genome without a reference; identifies error locations. | Requires high-coverage short-read data from the same individual. | High k-mer concordance; low error count. |
| Frameshift Analysis [60] | Presence of indels that disrupt coding sequences. | Direct evidence of assembly errors in protein-coding regions. | Relies on transcriptome data; underestimates errors in non-genic regions. | Few or no frameshifts in annotated genes. |
Evidence from plant genomics research underscores the importance of a multi-metric approach. For example, a study evaluating 41 genome assemblies of Triticeae crops (wheat, rye, and triticale) found that the proportion of complete BUSCO genes positively correlated with RNA-seq read mappability. Simultaneously, the frequency of internal stop codons in gene models served as a significant negative indicator of assembly accuracy [59]. This shows that BUSCO completeness and measures of correctness often align to identify high-quality assemblies.
Experimental Protocols for Orthogroup Conservation Analysis
The following workflow is adapted from methodologies used in recent plant single-cell transcriptomics and fungal genome assembly studies [5] [62]. It outlines a robust pipeline for using high-quality genomes in cross-species orthogroup analysis.
Detailed Experimental Methodology
Step 1: Genome Quality Assessment and Selection
Before comparative analysis, rigorously evaluate all candidate genome assemblies.
- BUSCO Analysis: Run BUSCO for each genome using a relevant lineage-specific dataset (e.g., "viridiplantae_odb10" for plants) in genome mode. The assembly should ideally achieve >95% complete BUSCO genes [61] [59].
- Correctness Check: Use a tool like Merqury to count assembly errors. Map short-reads from the same species to the assembly and calculate the single-nucleotide variant (SNV) rate, which serves as an error rate proxy [60].
- Contiguity Benchmark: Calculate contig and scaffold N50 statistics. Prioritize assemblies with high N50 for better gene model reconstruction [59] [60].
Step 2: Gene Prediction and Functional Annotation
- Employ an evidence-driven approach. Map available RNA-seq data (e.g., from PacBio Iso-Seq) to the genome to define accurate gene models [60].
- Annotate predicted genes using databases like UniRef90 and assign functional categories using Gene Ontology (GO) terms [62].
Step 3: Orthogroup Inference with OrthoFinder
- Provide the annotated protein sequences from all species being compared to OrthoFinder. This software infers orthogroups—sets of genes descended from a single gene in the last common ancestor [5].
- This step is critical for distinguishing between true orthologs and paralogs in subsequent analyses.
Step 4: Defining Orthologous Marker Groups (OMGs)
- This method, effective for identifying conserved cell types across plants, uses orthogroups as markers [5].
- For each cell cluster or tissue type in a reference species, identify its top N (e.g., 200) marker genes. Then, find all orthogroups that contain these marker genes. This collection of orthogroups constitutes the OMG for that cell type [5].
Step 5: Cross-Species Comparison and Validation
- Compare the OMGs from a query species against the reference database. Statistical tests like Fisher's exact test determine if the overlap in shared OMGs between clusters from different species is significant, indicating conserved cell identity or function [5].
- Validate findings with orthogonal data. For instance, conserved OMGs for meristematic cells might be enriched for GO terms related to ribosomal function and cell division, confirming their biological plausibility [5].
Impact of Assembly Quality on Orthogroup Analysis
The quality of the input genomes directly dictates the reliability of orthogroup conservation studies. Incomplete or incorrect assemblies can lead to both false-positive and false-negative conclusions.
- Fragmented Assemblies: Lead to fragmented gene models. During orthogroup inference with tools like OrthoFinder, a fragmented gene may not be assigned to any orthogroup or may be incorrectly classified, skewing the understanding of gene family evolution [59].
- Incorrect Assemblies (Frameshifts): Base errors that create frameshifts can make a true ortholog appear as a pseudogene, causing it to be excluded from analysis. This creates a false negative and an underestimation of conservation [60].
- Over-Duplication: High BUSCO duplication rates can indicate artificial duplication from assembly errors. This inflates the apparent size of gene families and creates false paralogs, complicating the identification of true one-to-one orthologs essential for precise cross-species mapping [5] [61].
Research demonstrates that robust genomes enable meaningful comparisons across vast evolutionary distances. For example, the OMG method successfully identified 14 dominant groups with conserved cell-type markers across 15 diverse plant species, including both monocots and dicots, by relying on high-quality assemblies and orthogroup definitions [5]. This would not be possible with poorly assembled genomes where orthologous relationships are obscured.
Validation and Comparative Insights: Case Studies Across Plant Lineages
A fundamental challenge in plant evolutionary biology is accurately identifying homologous cell types and gene functions across distantly related species. This is particularly complex when comparing monocots and dicots, which diverged approximately 200 million years ago and have since followed independent evolutionary trajectories. Conventional methods for cross-species cell type identification, which often rely on one-to-one orthologs or direct data integration, frequently fail in plants due to frequent gene duplication events and subsequent functional divergence [5] [40]. To address these limitations, a novel computational strategy termed Orthologous Marker Gene Groups (OMGs) has been developed, enabling robust cell identity determination and conservation analysis across diverse plant species, including deep comparisons between monocot and dicot lineages [5] [40]. This method represents a significant advancement in orthogroup conservation analysis by leveraging evolutionary relationships beyond simple sequence similarity, providing plant researchers with a powerful tool for comparative transcriptomics. This guide objectively examines the experimental performance of the OMG method in dicot-monocot comparisons, presenting supporting data and detailed methodologies to inform its application in plant research.
Understanding the OMG Method: Principles and Workflow
Conceptual Foundation
The OMG method addresses a critical bottleneck in plant single-cell RNA sequencing (scRNA-seq) analysis: the accurate annotation of cell types in non-model species where validated marker genes are scarce. Traditional approaches using one-to-one orthologous genes for cross-species integration prove unreliable in plants due to widespread gene family expansion from tandem duplication and whole-genome duplication events [5] [40]. The OMG method circumvents these limitations by using groups of orthologous genes as the fundamental unit for comparison, encompassing one-to-one, one-to-many, and many-to-many orthologous relationships that better represent the complex evolutionary history of plant genomes [5].
Computational Workflow
The OMG methodology follows a structured, three-stage workflow for systematic cross-species cell type identification:
- Stage 1: Marker Gene Identification - The top N marker genes (typically N=200) are identified for each cell cluster within a species using established tools such as Seurat [5] [40].
- Stage 2: Orthologous Group Construction - Orthologous gene groups across multiple plant species are generated using OrthoFinder software [5] [40].
- Stage 3: Statistical Comparison - Pairwise comparisons of overlapping OMGs between clusters in query and reference species are performed, with statistical significance assessed using Fisher's exact test and false discovery rate (FDR) correction [5] [40].
The following workflow diagram illustrates this process and its application to dicot-monocot comparisons:
Experimental Performance: Dicot-Monocot Validation
Comparative Analysis Framework
To validate its performance in dicot-monocot comparisons, the OMG method was tested using well-annotated single-cell datasets from Arabidopsis thaliana (dicot) and Oryza sativa (rice, monocot) roots [5]. The benchmark established 11 cell clusters in Arabidopsis and 10 in rice with corresponding cell types in the counterpart species [5]. Performance was quantified by the method's ability to correctly identify orthologous cell type pairs while minimizing false positives, with comparisons made against traditional one-to-one ortholog mapping approaches.
Quantitative Results and Performance Metrics
The OMG method demonstrated superior performance compared to conventional one-to-one ortholog mapping for dicot-monocot cell type identification:
Table 1: Performance Comparison of OMG vs. One-to-One Ortholog Methods in Dicot-Monocot Analysis
| Performance Metric | One-to-One Ortholog Method | OMG Method |
|---|---|---|
| Significant Cluster Pairs Identified | 8 pairs [5] | 14 pairs (FDR < 0.01) [5] |
| Orthologous Cell Type Pairs | 3 out of 8 pairs (37.5%) [5] | 13 out of 14 pairs (92.9%) [5] |
| Exact or Partial Matches | Not specified | 9 out of 14 pairs (64.3%) [5] |
| Mismatched Clusters | 5 out of 8 pairs (62.5%) [5] | 1 out of 14 pairs (7.1%) [5] |
The OMG method identified 14 statistically significant cluster pairs between Arabidopsis and rice, with 13 of these (92.9%) representing orthologous cell types [5]. Among these, 9 pairs were classified as exact or partial matches, indicating strong conservation of cellular identity between dicots and monocots [5]. The four clusters that showed mismatches were located in transitional zones on UMAP projections, suggesting they contain undifferentiated cells rather than representing true annotation errors [5]. Subsequent Gene Ontology analysis confirmed these clusters were enriched for ribosomal genes, a hallmark of meristematic cells, supporting this interpretation [5].
Extended Applications: Multi-Species Conservation Patterns
Broad Phylogenetic Conservation
The robustness of the OMG method enabled its application to a expansive dataset encompassing 15 diverse plant species, comprising 1 million cells organized into 268 cell clusters [5]. This large-scale analysis revealed 14 dominant cell identity groups with substantial marker conservation across both monocot and dicot lineages [5]. This finding demonstrates deep conservation of core cellular identities throughout angiosperm evolution, despite approximately 200 million years of independent evolution between these lineages.
Table 2: Conserved Cell-Type Groups Identified Through Multi-Species OMG Analysis
| Conserved Cell Group | Conservation Pattern | Functional Significance |
|---|---|---|
| Root Epidermal Cells | Conserved across monocots and dicots [5] | Interface with soil environment |
| Cortex Cells | Conservation with partial specialization [5] | Structural support and storage |
| Vascular Cell Types | Strong conservation [5] | Transport and mechanical support |
| Meristematic Cells | Highly conserved [5] | Growth and cell division |
| Leaf Mesophyll | Conserved with lineage-specific features | Photosynthesis |
| Guard Cells | Core conserved program [5] | Gas exchange regulation |
| Endodermis/Exodermis | Functional conservation [5] | Suberized barrier formation |
Detailed Experimental Protocols
OMG Analysis Workflow for Dicot-Monocot Comparisons
Researchers implementing the OMG method for cross-species comparisons should follow this detailed protocol:
Data Preprocessing and Cluster Marker Identification
- Process single-cell RNA-seq data for both dicot and monocot species using standard tools (e.g., Seurat v4 or later) [5] [40].
- Perform quality control, normalization, and clustering following best practices for plant single-cell data.
- Identify the top 200 marker genes for each cell cluster using appropriate statistical tests (e.g., Wilcoxon rank-sum test) with log fold change threshold of 0.25 [5].
Orthologous Group Construction
- Obtain protein sequences for all genes from both species and additional reference species as needed.
- Run OrthoFinder v2.0+ with default parameters to generate orthogroups across all species [5].
- Extract orthologous groups containing genes from both species of interest.
Cross-Species Comparison
- For each cluster in the query species (e.g., dicot), identify overlapping orthologous marker groups with each cluster in the reference species (e.g., monocot).
- Perform Fisher's exact test for each cluster pair comparison to determine statistical significance of OMG overlap.
- Apply multiple testing correction using Benjamini-Hochberg FDR procedure with significance threshold of FDR < 0.01 [5].
- Visualize results as a heatmap showing -log10(FDR) values for all cluster pairs.
Methodological Considerations
Several parameters require careful optimization for successful OMG analysis:
- Marker Gene Number: The selection of 200 marker genes per cluster represents a balanced parameter that provides sufficient overlapping OMGs while maintaining marker specificity. Smaller values (N<200) rapidly decrease overlapping markers, while larger values (N>200) reduce specificity [5].
- Evolutionary Distance: The method performs best when comparing species with appropriate evolutionary distances. For deep dicot-monocot comparisons, including intermediate species can improve orthogroup inference.
- Statistical Thresholds: The FDR threshold of 0.01 provides stringent control of false positives while identifying biologically meaningful conservation patterns [5].
Successful implementation of dicot-monocot comparisons using the OMG method requires the following key resources:
Table 3: Essential Research Reagents and Computational Tools for OMG Analysis
| Resource Category | Specific Tools/Resources | Application in OMG Workflow |
|---|---|---|
| Single-Cell Analysis | Seurat [5] [40], Scanpy | Data preprocessing, clustering, and marker gene identification |
| Orthology Inference | OrthoFinder [5] [40] | Construction of orthologous gene groups across species |
| Genome Annotations | Phytozome, Ensembl Plants | Gene models and functional annotations for reference genomes |
| OMG Implementation | OMG R Package [5], OMG Browser [5] | Specialized tools for OMG analysis and visualization |
| Reference Datasets | Arabidopsis single-cell atlas [5], Rice single-cell atlas [5] | Benchmarking and reference mapping |
The OMG method represents a significant advancement in cross-species comparative transcriptomics, specifically addressing the challenges posed by plant genome evolution. Through robust validation in dicot-monocot comparisons, this approach has demonstrated superior performance compared to conventional methods, correctly identifying orthologous cell types with high accuracy. The discovery of 14 conserved cell identity groups across 15 plant species reveals deep conservation of cellular programs despite extensive sequence divergence. For plant researchers investigating evolutionary biology, cell type identification, or comparative genomics, the OMG method provides a powerful framework for orthogroup conservation analysis that accounts for the complex evolutionary history of plant genomes.
The Asteraceae family (daisy or sunflower family) ranks among the largest angiosperm families, encompassing over 30,000 species that demonstrate remarkable adaptability to diverse and often extreme habitats worldwide [63] [64]. The genetic underpinnings of this ecological success have remained a central focus in evolutionary and plant biology. This guide objectively compares the established genomic and metabolic adaptations of Asteraceae, revealed through orthogroup conservation analysis, with metabolic systems prevalent in other plant families. We synthesize findings from foundational genomic comparisons and experimental data to provide a structured resource for researchers investigating the molecular basis of plant adaptation.
Central to this analysis is the discovery that Asteraceae have undergone a unique evolutionary path regarding their nitrogen-carbon (N-C) balance system, a core pathway governing nutrient assimilation and resource allocation in plants [63]. The following sections detail the genomic evidence for this rewiring, compare the system functionality against other plant models, and provide the methodological toolkit for replicating this type of large-scale comparative analysis.
Results and Data Comparison
Genomic Foundations of Asteraceae Divergence
Comparative genomics across 29 terrestrial plant species, including multiple Asteraceae and their close relatives, has uncovered significant evolutionary events shaping the family's lineage.
Table 1: Genomic Evolution Characteristics of Asteraceae
| Characteristic | Finding in Asteraceae | Implication for Adaptation |
|---|---|---|
| Origin & Polyploidization | Originated ~80 million years ago; experienced repeated paleopolyploidization [63] | Provided abundant genetic material for evolutionary innovation and trait diversification. |
| PII Gene Status | Conspicuous loss of the PII regulatory gene, which is otherwise near-universal in life [63] | Loss of a conserved master regulator, suggesting a fundamentally different N-C sensing mechanism. |
| Metabolic Gene Evolution | Stepwise upgrade of N-C system via gene duplications (e.g., for nitrogen uptake, fatty acid biosynthesis) [63] | Enhanced nitrogen uptake and carbon allocation to specific pathways like fatty acid synthesis. |
The Unique Nitrogen-Carbon Balance System
The defining metabolic adaptation in Asteraceae is the reconfigured Nitrogen-Carbon (N-C) balance system. In most plants, N and C metabolism is finely coordinated by sensor genes like PII, which acts as a reporter of the cellular energy state by binding effector molecules like ATP/ADP and 2-oxoglutarate (2-OG) [63]. Orthogroup analysis confirmed the conspicuous loss of the PII gene across the Asteraceae family, a regulator present in almost all other domains of life [63].
This loss is correlated with other genomic changes. The family has "upgraded" its N-C balance system through paleopolyploidization and tandem duplications of key metabolic genes, leading to a direct enhancement of nitrogen uptake and fatty acid biosynthesis pathways [63]. This unique system offers a potential molecular basis for the family's ecological success and a novel target for crop improvement strategies.
Table 2: Comparison of N-C Balance Systems in Plants
| System Component | Typical System (e.g., C3 Plants) | Asteraceae System |
|---|---|---|
| Core Regulator | PII protein present, senses C metabolic state (ATP/ADP, 2-OG) [63] | PII protein lost [63] |
| N Uptake & Assimilation | Nitrate absorbed, reduced to ammonium, assimilated via GS/GOGAT cycle [65] | Enhanced nitrogen uptake via duplicated metabolic genes [63] |
| C Allocation | Photoassimilates (sucrose) transported to sinks; starch as storage [65] | Enhanced fatty acid biosynthesis [63] |
| Response to Shading | In tea plants, shading promotes N metabolism (amino acids) and inhibits C metabolism (sugars) [66] | Not directly measured, but suggests N-C coordination can be environmentally modulated. |
Experimental Protocols
The insights into Asteraceae adaptation were derived from large-scale genomic comparisons. The following protocols detail the key methodologies.
Orthogroup Inference and Analysis
Principle: Orthogroup inference identifies sets of genes descended from a single gene in the last common ancestor of all species considered, providing a framework for comparative genomics [10].
Protocol:
- Data Collection: Assemble whole-genome sequencing data or transcriptomes (e.g., from databases like AGD, NCBI) for the target species and outgroups [63] [67] [68].
- Sequence Similarity Search: Perform an all-versus-all BLAST search of protein sequences across all species to obtain pairwise similarity scores [10].
- Score Normalization: Apply a normalization algorithm (e.g., as in OrthoFinder) to eliminate gene length and phylogenetic distance bias in BLAST scores. This step is critical for accuracy [10].
- Graph Construction: Build a graph where nodes represent genes, and edges represent normalized similarity scores between genes.
- Orthogroup Delimitation: Cluster the graph using an algorithm like MCL (Markov Cluster Algorithm) to infer discrete orthogroups [10].
- Downstream Analysis: Identify orthogroups that are expanded, conserved, or lost in specific lineages (e.g., Asteraceae). Perform functional enrichment analysis (GO, KEGG) on significant orthogroups [63] [68].
Identification of Genes Under Positive Selection
Principle: This analysis pinpoints genes with an excess of non-synonymous nucleotide substitutions (dN) over synonymous substitutions (dS), indicating adaptive evolution.
Protocol:
- Gene Alignment: For a target orthogroup, align the coding sequences (CDS) from multiple species.
- Phylogenetic Tree Construction: Build a species tree using single-copy orthologs or other conserved genes [68].
- Selection Test: Apply branch-site models within a framework like PAML (Phylogenetic Analysis by Maximum Likelihood). The model tests if the dN/dS ratio (ω) is significantly greater than 1 on a specific branch (e.g., the Asteraceae lineage).
- Statistical Validation: Use likelihood ratio tests to compare the model allowing positive selection with a null model that does not. Correct for multiple testing.
- Functional Interpretation: Annotate genes under positive selection to understand the traits under adaptive evolution (e.g., DNA repair, UV response, membrane transport in high-altitude Saussurea) [68].
Visualization of Concepts and Workflows
Orthogroup Analysis Workflow
The following diagram illustrates the computational pipeline for inferring orthogroups and analyzing their evolutionary dynamics, a key methodology underpinning the discovery of Asteraceae adaptations.
Figure 1: Orthogroup analysis workflow for multi-species genomic comparison.
Asteraceae N-C System Evolution
This diagram contrasts the standard plant N-C regulatory system with the unique model evolved in the Asteraceae family.
Figure 2: Comparative model of N-C balance system evolution in Asteraceae.
The Scientist's Toolkit
Table 3: Essential Research Resources for Asteraceae Genomics
| Resource Category | Specific Tool / Database | Function in Research |
|---|---|---|
| Genomics Databases | Asteraceae Genomics Database (AGD) [67] | Centralized platform for accessing curated Asteraceae genomes and associated omics data. |
| NCBI, GWH, 1K-MPGD [67] | Repositories for retrieving raw genomic and transcriptomic sequence data. | |
| Bioinformatics Software | OrthoFinder [10] | Accurate inference of orthogroups from whole-genome data, correcting for gene-length bias. |
| BUSCO [63] [68] | Assessment of genome/transcriptome assembly completeness based on universal single-copy orthologs. | |
| HMMER [69] | Protein domain analysis (e.g., identifying NTF2 and RRM domains in G3BP proteins). | |
| Molecular Biology Tools | Pfam, InterPro | Functional annotation of protein domains and families. |
| PAML (CodeML) [68] | Phylogenetic analysis by maximum likelihood, including tests for positive selection. |
The Oleaceae family, encompassing ecologically and economically vital genera such as Fraxinus (ash trees), Olea (olives), Jasminum (jasmine), Syringa (lilac), and Forsythia, represents a cornerstone of many terrestrial ecosystems and agricultural systems [70] [71]. A critical component of the plant immune system is the diverse family of intracellular immune receptors known as Nucleotide-binding leucine-rich repeat (NLR) genes. These genes encode proteins that recognize pathogen-derived effector molecules and initiate robust defense responses, a process known as Effector-Triggered Immunity (ETI) [70] [72]. The evolutionary dynamics of the NLR gene family—how it expands, contracts, and diversifies across species—are central to a plant's ability to adapt to evolving pathogenic threats. This guide provides a comparative analysis of NLR gene evolution across the Oleaceae family, framed within the context of orthogroup conservation. It objectively compares the distinct evolutionary strategies—conservation versus expansion—employed by different genera, supported by recent high-throughput genomic and transcriptomic data.
Orthogroup Conservation Analysis: A Framework for NLR Evolution
Orthogroup analysis, which clusters genes into groups descended from a single gene in a last common ancestor, is a powerful method for tracing gene family evolution across species. This approach allows researchers to identify core orthogroups conserved across lineages and lineage-specific orthogroups that have undergone expansion or loss, providing insights into evolutionary pressures and functional conservation [73].
In plant immunity, studying NLR orthogroups helps decipher how different species tailor their immune repertoires. A core orthogroup conserved across multiple genera suggests an essential, non-redundant function in the immune system, maintained over millions of years. In contrast, the expansion of specific orthogroups in a particular genus often indicates a recent, adaptive response to unique pathogenic pressures.
Table 1: Key Concepts in NLR Orthogroup Analysis
| Concept | Definition | Implication in Oleaceae Immunity |
|---|---|---|
| Orthogroup | A set of genes descended from a single gene in the last common ancestor of the species being compared [73]. | Serves as the fundamental unit for comparing NLR evolution across Fraxinus, Olea, and other genera. |
| Core Orthogroups | Orthogroups common to most or all the species studied, indicating conserved functions [73]. | Suggests retention of essential immune pathways; observed in Fraxinus with ancient Whole Genome Duplication (WGD)-derived NLRs. |
| Lineage-Specific Expansion | Significant increase in gene number within an orthogroup in a specific lineage. | Correlates with adaptation to new pathogens; prominently observed in the Olea genus [70]. |
| Gene Conservation | Evolutionary strategy characterized by low gene turnover and retention of ancestral genes. | Associated with specialized immune responses and potential energy efficiency, as seen in Fraxinus [70]. |
| Gene Birth-and-Death | Dynamic process involving frequent gene gains (duplications) and losses (pseudogenization) within a family [74]. | Shapes the NLR repertoire, leading to species-specific diversity, such as the pseudogenization of TIR-NLRs in Oleaceae [70]. |
Comparative Genomic Profiling of NLR Repertoires in Oleaceae
A high-throughput comparative genomic study analyzing 23 distinct Fraxinus species, alongside Olea europaea, Olea sylvestris, and other Oleaceae members, has revealed fundamental differences in how major genera manage their NLR immune arsenals [70].
The genus Fraxinus exhibits a predominant evolutionary strategy of gene conservation. Its NLR repertoire is largely shaped by the retention of genes acquired from an ancient whole-genome duplication (WGD) event approximately 35 million years ago (Mya) [70]. This strategy implies a system fine-tuned for specialized immune responses, potentially offering energy efficiency but possibly at the cost of flexibility against novel pathogens.
In stark contrast, the genus Olea (olives) has undergone extensive gene expansion. This is driven by recent duplication events and the emergence of novel NLR gene families, equipping olives with a highly diversified and potentially more adaptable immune receptor repertoire to recognize a broader array of pathogens [70]. Geographical adaptation, particularly in Old World ash species, has also contributed to dynamic patterns of gene expansion and contraction within the last 50 million years [70].
Table 2: Quantitative Comparison of NLR Gene Family Evolution in Select Oleaceae Genera
| Genus / Species | Primary Evolutionary Strategy | Key Genomic Events Shaping NLRs | Notable Patterns in NLR Subclasses |
|---|---|---|---|
| Fraxinus (Ash trees) | Gene Conservation [70] | Ancient WGD (~35 Mya) with widespread gene retention [70]. | Enhanced pseudogenization of TIR-NLRs; expansion of CCG10-NLRs [70]. |
| Olea (Olives) | Gene Expansion [70] | Recent duplications and significant birth of novel NLR families [70]. | Enhanced pseudogenization of TIR-NLRs; expansion of CCG10-NLRs [70]. |
| Oleaceae Family (All Species) | Mixed (Conservation & Expansion) | Varies by genus; influenced by geography and lineage-specific pressures [70]. | Universal trend of TIR-NLR pseudogenization and CCG10-NLR expansion across the family [70]. |
A universal trend across the Oleaceae family is the widespread pseudogenization (inactivation) of TIR-NLR (TNL) genes and a concurrent expansion of the CCG10-NLR subclass [70]. This indicates a major, family-wide shift in the architectural composition of the immune receptor repertoire.
Detailed Experimental Protocols for NLR Gene Analysis
The insights into Oleaceae NLR evolution are derived from sophisticated bioinformatic and genomic workflows. The following protocols detail the key methodologies used in the cited research.
Protocol 1: Genome-Wide Identification and Annotation of NLR Genes
This protocol is designed for the high-throughput mining of NLR genes from genomic data [70] [74].
- Data Acquisition: Obtain publicly available genomic resources, including genome assemblies and annotated proteomes, from repositories such as NCBI, CuGenDB, or Phytozome [70] [74].
- NLR Gene Mining: Subject the proteome files to the NLRtracker pipeline or a similar tool. This pipeline uses predefined rules to identify proteins containing characteristic NLR domains [70].
- Domain Verification and Classification: Confirm the identity of candidate NLR genes by scanning them against hidden Markov models (HMMs) of known domains (e.g., the NB-ARC domain, Pfam ID: PF00931) using tools like HMMER3 [74] [75]. Classify genes into subclasses (TNL, CNL, RNL) based on their N-terminal domains (TIR, CC, or RPW8) [75].
- Motif Analysis: Use the MEME (Multiple EM for Motif Elicitation) suite to identify conserved amino acid motifs within the NB-ARC domain of the identified NLRs [74] [75].
- Orthogroup Delineation: Cluster the identified NLR genes from multiple species into orthogroups using a tool like OrthoFinder, which employs algorithms such as DIAMOND for sequence similarity and MCL for clustering [73].
The workflow for this protocol is summarized in the diagram below.
Protocol 2: Evolutionary and Expression Analysis of NLR Genes
This protocol builds on the identified NLRs to understand their evolution and functional expression [70] [73] [75].
- Phylogenetic Analysis: Extract the NBS domain sequences from the classified NLR genes. Perform multiple sequence alignment using ClustalW or MAFFT. Construct a phylogenetic tree with the Maximum Likelihood method using software like IQ-TREE [75].
- Analysis of Gene Dynamics: Reconcile the NLR gene tree with the known species tree using software like NOTUNG to infer historical gene duplication and loss events [75].
- Transcriptomic Validation: Retrieve RNA-seq datasets from public archives like the Sequence Read Archive (SRA). Map the reads to the reference genome and quantify gene expression levels (e.g., in FPKM or TPM) [70] [73].
- Differential Expression: Analyze expression data to compare NLR transcript levels across different conditions (e.g., susceptible vs. tolerant cultivars, infected vs. mock-treated tissues) to associate specific NLRs with immune responses [73].
- Functional Validation (VIGS): For candidate genes, perform functional validation using Virus-Induced Gene Silencing (VIGS). This involves cloning a fragment of the candidate NLR into a VIGS vector, inoculating plants, and then challenging them with a pathogen to observe if susceptibility increases [73].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Cutting-edge research in plant immuno-genomics relies on a suite of bioinformatic tools, genomic resources, and experimental reagents.
Table 3: Key Research Reagent Solutions for NLR Gene Analysis
| Reagent / Resource | Type | Function in NLR Research |
|---|---|---|
| NLRtracker Pipeline | Bioinformatics Software | Automated, high-throughput identification and annotation of NLR genes from genomic or transcriptomic data [70]. |
| OrthoFinder | Bioinformatics Software | Infers orthogroups and gene families from whole proteome sequences, crucial for comparative evolutionary analysis [73]. |
| AlphaFold / AI Tools | Computational Model | Predicts the 3D structure of proteins, enabling rational engineering of immune receptors like FLS2 and NLRs for broader pathogen recognition [76] [77]. |
| VIGS (Virus-Induced Gene Silencing) Kit | Functional Validation Reagent | Knocks down the expression of a candidate NLR gene in planta to rapidly test its function in disease resistance [73]. |
| SRA RNA-seq Datasets | Genomic Data | Provides transcriptomic evidence for NLR gene expression under various biotic and abiotic stresses [70] [73]. |
| Chromosome-Level Genome Assemblies | Genomic Data | Essential for accurately determining the genomic location, arrangement, and cluster analysis of NLR genes [70]. |
This comparison guide delineates the two dominant evolutionary patterns shaping the NLR immune arsenal in the Oleaceae family: the conservative strategy of Fraxinus, which maintains ancient, specialized immune genes, and the expansive strategy of Olea, which rapidly diversifies its receptor repertoire. These patterns, revealed through orthogroup conservation analysis, highlight how closely related species can adopt divergent paths to immune adaptation.
The future of harnessing NLR genes for crop improvement is moving toward synthetic biology. Instead of simply transferring NLRs between species, researchers are now engineering them for enhanced function. Approaches include creating "pikobodies" by swapping NLR recognition domains with nanobodies to target specific pathogen effectors and using AI-powered tools like AlphaFold to rationally design receptors that recognize a wider spectrum of pathogens [76] [77]. These strategies, combined with a deeper understanding of NLR evolution as revealed in studies of families like Oleaceae, promise to revolutionize our ability to design durable disease resistance in plants.
Orthogroup inference, the process of identifying sets of genes descended from a single ancestral gene in a common ancestor, forms the foundational step in comparative genomics [38]. In plant biology, accurate orthogroup identification is crucial for investigating cell-type conservation, gene function, and evolutionary relationships across species [40]. However, this task presents significant challenges due to gene family expansions, frequent tandem duplications, and whole-genome duplication events that are particularly common in plant lineages [40]. These complexities have driven the development of various computational methods for orthogroup analysis, which generally fall into two categories: traditional integration-based approaches and the more recently developed Orthologous Marker Gene Groups (OMG) method.
This guide provides a comprehensive performance comparison between these competing methodologies, focusing on their application in plant species research. We examine quantitative performance metrics, detail experimental protocols, and provide practical resources to enable researchers to select the optimal approach for their specific research objectives, particularly in the context of drug development from medicinal plants where understanding biosynthetic pathways across species is critical [78] [79].
Performance Benchmarking: Quantitative Comparison
Rigorous benchmarking reveals significant differences in the performance characteristics between the OMG method and traditional integration approaches. The table below summarizes key performance metrics based on controlled experiments using plant single-cell RNA sequencing (scRNA-seq) data:
Table 1: Performance comparison between OMG and traditional integration methods
| Performance Metric | OMG Method | Traditional Integration Methods |
|---|---|---|
| Cell-type identification accuracy | 13/14 orthologous cell types identified between Arabidopsis and rice [40] | 3/8 orthologous cell types identified using one-to-one orthologs [40] |
| Computational requirements | Lower (no data integration required) [40] | Significant (requires constructing and iterating gene-gene bipartite graphs) [40] |
| Cross-species scalability | High (successfully mapped 1 million cells, 268 clusters across 15 species) [40] | Limited by computational constraints of integration [40] |
| Handling of gene family expansions | Robust (accounts for one-to-one, one-to-many, and many-to-many orthologs) [40] | Problematic (fails with one-to-one orthologs due to sequence similarity limitations) [40] |
| Methodology | Marker-based using Orthologous Marker Gene Groups [40] | Integration-based using one-to-one orthologous genes [40] |
The Orthobench benchmark, which contains 70 expert-curated reference orthogroups (RefOGs), provides a standardized framework for evaluating orthogroup inference accuracy [38]. When applied to this benchmark, the OMG method demonstrates superior performance in handling the complex evolutionary relationships characteristic of plant genomes.
Methodological Approaches: Experimental Protocols
OMG Method Workflow
The Orthologous Marker Gene Groups (OMG) method employs a structured, statistics-based approach for cell-type identification and orthogroup analysis:
- Marker Gene Identification: The top N marker genes (typically N=200) are identified for each cell cluster in each species using established tools such as Seurat [40].
- Orthogroup Generation: OrthoFinder or similar tools are used to generate orthologous gene groups across multiple plant species (M=15 in the reference study) [40].
- Pairwise Comparison: Statistical tests (Fisher's exact test) are performed to identify clusters with significant numbers of shared OMGs between query and reference species [40].
- Significance Thresholding: Results are filtered using false discovery rate (FDR) correction (FDR < 0.01) to determine biologically meaningful matches [40].
The optimal marker gene number (N=200) was determined empirically - values below 200 caused rapid decrease in overlapping markers across species, while higher values reduced specificity [40].
OMG Method Computational Workflow
Traditional Integration Approaches
Traditional integration-based methods employ fundamentally different strategies for cross-species comparison:
- Data Integration: Single-cell data from diverse species are integrated using one-to-one orthologous genes as anchors [40].
- Manifold Projection: Methods such as SAMap project datasets into a shared manifold space by creating gene-by-gene bipartite graphs and iteratively refining them [40].
- Co-expression Proxies: Some improved methods identify one-to-one gene pairs called co-expression proxies to provide better anchors for integration [40].
- Label Transfer: Cell types in the integrated data are determined by transferring annotations from reference species [40].
A significant limitation of these approaches is their reliance on one-to-one orthologous genes, which are assigned solely based on sequence similarity without accounting for potential divergence in cell type-specific expression patterns [40].
Traditional Integration Method Workflow
Experimental Validation and Case Studies
Cross-Species Cell Type Identification
The performance difference between methods was quantitatively evaluated through a controlled experiment comparing cell type identification accuracy between Arabidopsis (dicot) and rice (monocot) roots:
Table 2: Cross-species cell type identification accuracy between Arabidopsis and rice
| Method | Significant Cluster Pairs Identified | Orthologous Cell Type Matches | Mismatched/Undifferentiated Clusters |
|---|---|---|---|
| OMG Method | 14 pairs (FDR < 0.01) [40] | 13/14 pairs from orthologous cell types (9 exact/partial matches) [40] | 4 clusters in connecting regions of UMAP [40] |
| Traditional Integration | 8 pairs using one-to-one orthologs [40] | 3/8 pairs from orthologous cell types [40] | Not specified |
The OMG method demonstrated superior accuracy in identifying orthologous cell types, correctly matching 13 out of 14 significant cluster pairs compared to only 3 out of 8 using traditional integration with one-to-one orthologs [40]. Mismatched clusters in the OMG method were primarily found in connecting regions of UMAP visualizations, suggesting these represent undifferentiated cells rather than methodological errors [40].
Handling of Evolutionary Divergence
The OMG method was specifically designed to address challenges in plant genomics that limit traditional approaches:
- Gene Family Expansions: Plants frequently undergo tandem duplication and whole-genome duplication, creating one-to-many and many-to-many orthologous relationships that violate the one-to-one assumption of traditional methods [40].
- Marker Expression Divergence: Closest orthologs of cell-type identity marker genes in reference species like Arabidopsis often don't exhibit the same cell-type specificity across diverse plant species [40].
- Computational Efficiency: The OMG approach doesn't require cross-species data integration, making it faster and more scalable for comparisons across multiple species [40].
Research Reagent Solutions
Implementing these computational methods requires specific tools and resources. The following table details essential research reagents and computational tools for orthogroup conservation analysis:
Table 3: Essential research reagents and computational tools for orthogroup analysis
| Tool/Resource | Function | Application Context |
|---|---|---|
| OrthoFinder [40] [38] | Infers orthogroups from genomic data | Generation of orthologous gene groups across multiple species |
| Seurat [40] | Identifies marker genes from single-cell data | Initial marker gene identification for cell clusters |
| Single-cell RNA sequencing data [40] | Profiles gene expression at single-cell resolution | Cell type identification and comparison across species |
| OMG Browser [40] | User-friendly web-based tool for cell-type identification | Simplifies OMG method application for plant datasets |
| Orthobench Benchmark [38] | Standardized benchmark with 70 reference orthogroups | Evaluation of orthogroup inference accuracy |
| MAFFT [38] | Multiple sequence alignment algorithm | Alignment of gene sequences for phylogenetic analysis |
| IQ-TREE [38] | Phylogenetic tree inference | Gene tree construction for orthogroup delineation |
Implications for Drug Development Research
The enhanced accuracy of orthogroup identification provided by the OMG method has significant implications for drug development from medicinal plants:
- Biosynthetic Pathway Conservation: Identifying conserved orthogroups across species helps researchers trace the evolutionary history of biosynthetic pathways for valuable secondary metabolites with therapeutic potential [78].
- Cross-Species Transcriptome Analysis: Accurate cell-type identification enables more precise comparison of specialized cell types producing bioactive compounds across medicinal plant species [40] [78].
- Metabolic Engineering: Correct orthogroup assignment facilitates the identification of key enzymes in biosynthetic pathways, supporting metabolic engineering approaches to enhance production of valuable compounds [78].
Multiomics technologies, including metabolomics, proteomics, and transcriptomics, are revolutionizing our ability to characterize and enhance the production of valuable secondary metabolites from traditional medicinal plants [78]. The application of accurate orthogroup analysis methods like OMG enhances these approaches by providing reliable cross-species comparisons essential for understanding the evolution and distribution of phytochemical pathways.
The emergence of single-cell RNA sequencing (scRNA-seq) has revolutionized the study of plant development and biology, enabling the characterization of cellular heterogeneity at an unprecedented resolution. A significant challenge in plant single-cell biology, however, has been the accurate identification of cell types across different species and the ability to investigate the conservation of these cell types throughout plant evolution. The scarcity of known cell-type marker genes and the divergence of marker expression patterns have traditionally limited our capacity to identify cell types accurately and perform cross-species comparisons in many plant species [50].
Addressing this challenge requires computational strategies that can bridge the gap between uncharacterized scRNA-seq datasets and prior biological knowledge. The interpretation of scRNA-seq data is often manual and represents a common bottleneck in analysis workflows, as even the concept of a "cell type" lacks a clear, computationally amenable definition, with most practitioners relying on an intuitive, "I'll know it when I see it" approach [80]. This problem is particularly acute in cross-species comparisons, where differences in genome organization and gene expression patterns between monocots and dicots—the two major angiosperm subclasses that diverged from common ancestors approximately 200 million years ago—can complicate comparative analyses [81] [82].
Against this backdrop, a novel computational strategy called Orthologous Marker Gene Groups (OMGs) has been developed to identify cell types in both model and non-model plant species and enable rapid comparison of cell types across published single-cell maps [50]. This method does not require cross-species data integration while still accurately determining inter-species cellular similarities, providing a powerful framework for exploring the depth of cell-type conservation across the monocot-dicot divide.
Orthologous Marker Gene Groups (OMGs): Methodology and Workflow
Core Computational Framework
The OMG method represents a significant advancement in computational biology for cross-species cell-type identification. Its design specifically addresses the limitations of traditional marker-based annotation and reference-based integration approaches. The methodology operates through several key stages:
Orthologous Gene Group Construction: The initial phase involves identifying groups of orthologous genes across the species of interest. This establishes an evolutionarily conserved framework for comparison that respects the genetic divergence between monocots and dicots, which exhibit approximately 80% difference in their coding sequences despite sharing a core set of biological functions [81].
Conserved Marker Gene Identification: For each putative cell type, the method identifies marker genes whose expression patterns are conserved across species. This step moves beyond simple sequence homology to incorporate expression pattern conservation, which is crucial for meaningful biological comparison.
Cellular Similarity Quantification: The method computes similarities between cells from different species based on the expression of orthologous marker gene groups, enabling the identification of conserved cell types without requiring data integration that might distort biological signals.
A key advantage of the OMG approach is its ability to function without requiring cross-species data integration, while still accurately determining inter-species cellular similarities [50]. This is particularly valuable when comparing monocots and dicots, which despite their shared ancestry, exhibit significant genomic differences including variations in genome size, transposable element content, and microsatellite distribution [82].
Experimental Validation and Performance
The OMG method has been rigorously validated through analysis of published single-cell data from species with well-annotated single-cell maps. The validation demonstrated that the method can capture the majority of manually annotated cell types with high accuracy [50]. Furthermore, the robustness of the method was tested through its application to a massive dataset comprising 1 million cells and 268 cell clusters across 15 diverse plant species, representing one of the most comprehensive cross-species single-cell analyses in plants to date [50].
Table 1: Key Features of the OMG Methodology
| Feature | Description | Advantage |
|---|---|---|
| Reference-free Design | Does not require expertly annotated reference datasets | Applicable to non-model species |
| Orthology-Based Comparison | Uses evolutionarily conserved gene groups | Respects genetic divergence between species |
| Scalability | Successfully applied to 1 million cells across 15 species | Suitable for large-scale atlas projects |
| Accessibility | Available through user-friendly OMG browser | Accessible to biologists without computational expertise |
Key Finding: 14 Conserved Cell-Type Groups Across Monocots and Dicots
The Conservation Landscape
The application of the OMG method to the extensive dataset of 15 plant species revealed a remarkable degree of cell-type conservation between monocots and dicots. The analysis identified 14 dominant groups with substantial conservation in shared cell-type markers across these evolutionarily divergent lineages [50]. This finding indicates that despite approximately 200 million years of independent evolution following the monocot-dicot divergence, fundamental cellular identities have been maintained throughout angiosperm evolution.
The discovery of these 14 conserved cell-type groups represents a significant breakthrough in our understanding of plant evolution and development. It suggests that the genetic programs defining these core cellular identities were already established in the common ancestor of monocots and dicots and have been under strong evolutionary constraint ever since. This conservation persists despite the extensive genomic differences between monocots and dicots, including variations in genome size, transposable element content, and microsatellite distribution patterns [82].
Methodological Advantages for Cross-Species Comparison
The OMG method provides several distinct advantages for identifying these conserved cell-type groups compared to traditional approaches:
Avoidance of Integration Artifacts: By not requiring data integration across species, the method prevents technical artifacts that might obscure true biological conservation signals.
Resolution of Evolutionary Relationships: The orthology-based approach respects the evolutionary divergence between monocots and dicots while still identifying deeply conserved cellular programs.
Applicability to Non-Model Species: The method enables cell-type identification in species without existing well-annotated single-cell maps, facilitating the study of cellular conservation across the full diversity of angiosperms.
These technical advantages make the OMG method particularly suited for exploring the depth of cellular conservation between monocots and dicots, whose genomes exhibit both shared synteny and significant reorganization since their divergence [81].
Comparative Context: Genomic Conservation Between Monocots and Dicots
Structural and Functional Genomic Conservation
The discovery of 14 conserved cell-type groups between monocots and dicots aligns with broader patterns of genomic conservation identified through comparative genomics. Studies of ABCB1 genes, which encode auxin transporters critical for plant development, reveal significant structural and functional conservation between monocots and dicots despite sequence divergence [83].
Table 2: Comparative Genomics of Monocots and Dicots
| Genomic Feature | Monocots | Dicots | Conservation Pattern |
|---|---|---|---|
| ABCB1 Gene Size | Average ~5.8 kb | Average ~5.7 kb | High structural conservation |
| Intron-Exon Junctions | Conserved | Conserved | Near perfect conservation |
| Nucleotide Binding Domains | Highly conserved | Highly conserved | Functional constraint |
| Microsatellite Distribution | 204.92 SSR loci/Mb | 451.26 SSR loci/Mb | Differential abundance |
| Coding Region SSRs | 144.45/Mb | 67.52/Mb | Higher in monocots |
Research on ABCB1 orthologs demonstrates that the sequence identity of true orthologs ranges from 56-90% at the DNA level and 75-91% at the amino acid level, with conserved intron-exon junctions across species [83]. The predicted 3D protein structures of these transporters show remarkable similarity between monocots and dicots, particularly for residues involved in auxin binding, indicating strong functional conservation despite sequence divergence [83].
Genome Size and Trait Relationships
Recent phylogenetically informed analyses of angiosperm genomes have revealed that the interaction between life cycle (annual vs. perennial) and monocot-dicot distinction is a primary determinant of genome size variation, with perennial monocots exhibiting the largest genome sizes [84] [85]. These studies examining 2,285 angiosperm species found that patterns of correlation between genome size and functional traits are often group-specific and sometimes reversed between monocots and dicots, reflecting divergent adaptive strategies.
For example, genome size (1C value) shows a positive correlation with plant height in annuals but a negative correlation in perennials [84] [85]. After phylogenetic correction using Phylogenetic Generalized Least Squares (PGLS) analyses, some associations disappear while others remain robust, indicating a mix of ancestral constraints and adaptive significance in genome size-trait relationships [84] [85].
Experimental Protocols and Research Workflows
Single-Cell RNA Sequencing and Analysis Pipeline
The identification of conserved cell types across monocots and dicots relies on standardized single-cell RNA sequencing workflows. A typical scRNA-seq analysis pipeline involves several key stages [86] [87]:
Sample Preparation and Single-Cell Isolation: Tissue dissociation to create single-cell suspensions from plant organs of interest.
Library Preparation and Sequencing: Using platforms such as 10x Genomics to generate barcoded single-cell libraries for high-throughput sequencing.
Quality Control and Filtering: Removing low-quality cells, doublets, and cells with high mitochondrial or chloroplast gene content.
Normalization and Batch Correction: Accounting for technical variation and batch effects to ensure cells cluster by biological similarity rather than technical artifacts.
Dimensionality Reduction and Clustering: Using PCA, UMAP, or t-SNE to visualize cells in two dimensions and clustering algorithms to group cells with similar expression profiles.
Cell Type Annotation: Applying methods like OMG, Azimuth, or marker-based annotation to identify cell types.
Cross-Species Comparison: Using orthology-based methods to identify conserved cell types across evolutionary distances.
Orthologous Marker Group Identification Workflow
Figure 1: Orthologous Marker Groups (OMG) Computational Workflow
Computational Tools for Cell-Type Annotation
Table 3: Essential Research Tools for Cross-Species Cell-Type Analysis
| Tool/Resource | Type | Function | Application Context |
|---|---|---|---|
| OMG Browser | Web Tool | Cell-type identification using Orthologous Marker Groups | Cross-species cell-type comparison |
| Azimuth | Automated Annotation Tool | Reference-based cell-type annotation using predefined references | Tissue-specific annotation within species |
| SingleR | Computational Method | Reference-based annotation without predefined markers | Comparison to reference datasets |
| scType | Marker-Based Tool | Cell-type identification using marker gene lists | Rapid annotation of common cell types |
| Seurat | Analysis Toolkit | End-to-end scRNA-seq analysis including clustering and visualization | Primary data analysis and visualization |
| Loupe Browser | Visualization Software | Interactive exploration of single-cell data from 10x Genomics | Data sharing and collaborative analysis |
Practical Implementation Guidance
For researchers implementing these methods, several practical considerations are essential:
Reference Dataset Selection: When using reference-based annotation tools like Azimuth, the choice of reference dataset significantly impacts annotation accuracy. Selecting a reference that appropriately matches the tissue type and species of interest is crucial [88].
Subclustering Strategy: For resolving fine-grained cellular heterogeneity, a subclustering approach is recommended, where initially identified clusters are subset and re-analyzed to identify more specific cell subtypes or states [87].
Multi-Method Validation: Given that no single annotation method is perfect, employing multiple complementary approaches and seeking consensus between them increases confidence in annotation results [87].
Orthology Quality: The accuracy of OMG-based comparisons depends heavily on the quality of orthology predictions, requiring careful curation of orthologous gene groups, particularly for rapidly evolving gene families.
The identification of 14 dominant cell-type groups with substantial conservation between monocots and dicots provides profound insights into the evolution of plant development. This conservation suggests that fundamental genetic programs controlling cellular identity were established early in angiosperm evolution and have been maintained under strong selective pressure despite approximately 200 million years of independent evolution.
These findings have significant implications for plant biotechnology and crop improvement. The conserved nature of these cell-type identities means that biological insights gained in model systems may be more broadly applicable across angiosperms than previously expected. Furthermore, the OMG method and associated resources provide a powerful framework for studying cellular function and development across a wide range of plant species, facilitating the transfer of knowledge from well-characterized model systems to less-studied crop species.
As single-cell technologies continue to advance and are applied to an ever-growing diversity of plant species, our understanding of the conservation and diversification of cell types throughout plant evolution will continue to deepen. The discovery of these 14 conserved cell-type groups represents just the beginning of this exciting journey into the cellular basis of plant diversity and evolution.
Conclusion
Orthogroup conservation analysis represents a paradigm shift in comparative plant genomics, providing robust frameworks for identifying evolutionarily conserved gene networks and cellular functions across diverse species. The integration of methods like Orthologous Marker Groups and OrthoFinder has demonstrated remarkable success in revealing 14 conserved cell-type groups across monocots and dicots, uncovering specialized adaptation systems in Asteraceae, and tracing immune gene evolution in Oleaceae. These findings not only advance fundamental plant biology but offer valuable models for biomedical research, particularly in understanding how conserved genetic networks underlie specialized metabolic functions and defense mechanisms. Future directions should focus on expanding orthogroup databases, integrating single-cell multi-omics data, and developing translational applications that leverage plant evolutionary innovations for biomedical advances, including drug discovery and understanding conserved cellular resilience mechanisms.
References
- 1. Obituary: Walter Fitch and the orthology paradigm - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3178060/]
- 2. Orthologs and paralogs - we need to get it right [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2001-2-8-interactions1002]
- 3. Functional and evolutionary implications of gene orthology [https://pmc.ncbi.nlm.nih.gov/articles/PMC5877793/]
- 4. PlantOrDB: a genome-wide ortholog database for land plants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4481079/]
- 5. Orthologous marker groups reveal broad cell identity ... [https://www.nature.com/articles/s41467-024-55755-0]
- 6. Testing the Ortholog Conjecture with Comparative Functional ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1002073]
- 7. Conservation and Remodeling of Alternative Splicing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12609446/]
- 8. Editorial: Plant-derived natural compounds in drug discovery [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.1042695/full]
- 9. Orthologs of Human-Disease-Associated Genes in Plants Are ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9965218/]
- 10. OrthoFinder: solving fundamental biases in whole genome... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0721-2]
- 11. OMA orthology database: Types of orthologous groupings [https://omabrowser.org/oma/homologs/]
- 12. A multilayered cross- species of GRAS transcription factors... analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC8020295/]
- 13. OrthoFinder: phylogenetic orthology inference for comparative genomics | Genome Biology | Full Text [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1832-y]
- 14. GitHub - davidemms/OrthoFinder: Phylogenetic orthology inference for comparative genomics [https://github.com/davidemms/OrthoFinder]
- 15. (PDF) The Plant Orthology Browser: An Orthology and Gene‐Order... [https://www.academia.edu/104601547/The_Plant_Orthology_Browser_An_Orthology_and_Gene_Order_Visualizer_for_Plant_Comparative_Genomics]
- 16. Analysis of 41 plant genomes supports a wave ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4120086/]
- 17. Review Plant evolutionary genomics [https://www.sciencedirect.com/science/article/abs/pii/S136952660100231X]
- 18. Phylogenomic profiles of whole-genome duplications in ... [https://www.nature.com/articles/s41467-024-47428-9]
- 19. The impact of whole genome duplications on the human gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8675932/]
- 20. AgroLD: a knowledge graph for the plant sciences [https://bmcgenomdata.biomedcentral.com/articles/10.1186/s12863-025-01359-6]
- 21. An integrative 3D-genome database for plants - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12177499/]
- 22. Comparative analysis, diversification, and functional ... [https://www.nature.com/articles/s41598-024-62876-5]
- 23. OrthoQuantum: visualizing evolutionary repertoire of eukaryotic... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9252792/]
- 24. plant genes responsive to root-knot... Evolutionarily conserved [https://link.springer.com/article/10.1007/s00438-020-01677-7]
- 25. Comparative genomics and evolutionary analysis of plant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9400807/]
- 26. FastOMA: a fast and accurate orthology inference tool [https://communities.springernature.com/posts/fastoma-a-fast-and-accurate-orthology-inference-tool]
- 27. Different orthology inference algorithms generate similar ... [https://pubmed.ncbi.nlm.nih.gov/39906489/]
- 28. 4. OrthoFinder best practices [https://davidemms.github.io/orthofinder_tutorials/orthofinder-best-practices.html]
- 29. 3. Exploring OrthoFinder's results [https://davidemms.github.io/orthofinder_tutorials/exploring-orthofinders-results.html]
- 30. Repository for identification of orthologous marker genes in ... [https://github.com/LiLabAtVT/OrthoMarkerGeneGroups]
- 31. PlantTribes2: Tools for comparative gene family analysis in ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.1011199/full]
- 32. PlantTribes2: Tools for comparative gene family analysis in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9928214/]
- 33. Building a foundation for gene family analysis in Rosaceae ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.975942/full]
- 34. PlantTribes: a gene and gene family resource for comparative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2238917/]
- 35. PlantTribes - Database Commons [https://ngdc.cncb.ac.cn/databasecommons/database/id/1432]
- 36. dePamphilis/PlantTribes [https://github.com/dePamphilis/PlantTribes]
- 37. Orthogroup and phylotranscriptomic analyses identify ... [https://www.sciencedirect.com/science/article/pii/S2590346223002122]
- 38. Benchmarking Orthogroup Inference Accuracy [https://pmc.ncbi.nlm.nih.gov/articles/PMC7738749/]
- 39. A multi-omics and taxonomy database for woody plants - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010388/]
- 40. Orthologous marker groups reveal broad cell identity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11695714/]
- 41. Plant Single-Cell Cell Type Annotation with the OMG Browser [https://www.omicsempower.com/blog/plant-single-cell-cell-type-annotation-omg-browser/]
- 42. Recent progress in single-cell transcriptomic studies in plants [https://link.springer.com/article/10.1007/s11816-025-00967-z]
- 43. Single-cell omics in plant biology - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12214168/]
- 44. A single-cell, spatial transcriptomic atlas of the Arabidopsis ... [https://www.nature.com/articles/s41477-025-02072-z]
- 45. Using all Gene Families Vastly Expands Data Available for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9178227/]
- 46. Gene Family Expansions Provide Molecular Flexibility ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12453137/]
- 47. Unique trajectory of gene family evolution from genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11185758/]
- 48. A long road ahead to reliable and complete medicinal plant ... [https://www.nature.com/articles/s41467-025-57448-8]
- 49. Harnessing plant biosynthesis for the development of next ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3002886]
- 50. Orthologous marker groups reveal broad cell identity ... [https://pubmed.ncbi.nlm.nih.gov/39747890/]
- 51. Tissue-Specificity of Gene Expression Diverges Slowly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5193323/]
- 52. Benchmarking ortholog identification methods using functional ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2006-7-4-r31]
- 53. False Discovery Rate [https://www.publichealth.columbia.edu/research/population-health-methods/false-discovery-rate]
- 54. False discovery rate [https://en.wikipedia.org/wiki/False_discovery_rate]
- 55. Gene duplication and evolution in recurring polyploidization ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1650-2]
- 56. The application conditions of false discovery rate control [https://pmc.ncbi.nlm.nih.gov/articles/PMC10311093/]
- 57. A curated benchmark dataset for molecular identification ... [https://www.nature.com/articles/s41597-025-05230-2]
- 58. An integrative 3D-genome database for plants [https://www.sciencedirect.com/science/article/pii/S2590346225001087]
- 59. Evaluating Genome Assemblies for Optimized ... [https://www.mdpi.com/2223-7747/14/7/1140]
- 60. assessing the quality of genome assemblies with the 3 Cs [https://www.pacb.com/blog/beyond-contiguity/]
- 61. Genome Completeness Assessment with BUSCO [https://www.biobam.com/genome-completeness-assessment-with-busco/]
- 62. Evaluating the Assembly Strategy of a Fungal Genome from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12387558/]
- 63. Comparative genomics reveals a unique nitrogen-carbon ... [https://www.nature.com/articles/s41467-023-40002-9]
- 64. Adaptive convergence and divergence underpin the ... [https://www.sciencedirect.com/science/article/abs/pii/S0367253024001075]
- 65. Recent Advances in Carbon and Nitrogen Metabolism in C3 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7795015/]
- 66. Carbon and Nitrogen Metabolism Are Jointly Regulated ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.894840/full]
- 67. a comprehensive platform for Asteraceae genomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11366637/]
- 68. Transcriptomes of Saussurea (Asteraceae) Provide ... [https://www.mdpi.com/2223-7747/10/8/1715]
- 69. Comprehensive evolutionary analysis and nomenclature of ... [https://www.life-science-alliance.org/content/5/9/e202101328]
- 70. Tracing the path from conservation to expansion evolutionary ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-06233-2]
- 71. Tracing the path from conservation to expansion evolutionary ... [https://agris.fao.org/search/en/providers/122436/records/67dac655677d8be0233bcb0a]
- 72. Evolution and Conservation of Plant NLR Functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3782705/]
- 73. Comparative analysis , diversification, and functional validation of plant ... [https://www.nature.com/articles/s41598-024-62876-5?error=cookies_not_supported&code=95da165c-75ff-42d2-9bbb-aa8142196912]
- 74. Large-scale gene gains and losses molded the NLR defense arsenal... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8463517/]
- 75. Comparative-genomic analysis reveals dynamic NLR gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10017999/]
- 76. Rewriting the code of plant immunity | Nature Biotechnology [https://www.nature.com/articles/s41587-025-02886-4]
- 77. How AI is supercharging plant immunity to fight deadly ... [https://www.sciencedaily.com/releases/2025/07/250730030349.htm]
- 78. Revisiting Traditional Medicinal Plants: Integrating Multiomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12252194/]
- 79. Medicinal plants meet modern biodiversity science [https://www.sciencedirect.com/science/article/pii/S0960982223017384]
- 80. Chapter 7 Cell | Basics of Single-Cell Analysis with... type annotation [https://www.bioconductor.org/books/3.13/OSCA.basic/cell-type-annotation.html]
- 81. Comparative genome analysis of monocots and dicots ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166904000308]
- 82. Comparative genomics analysis gives insights into ... [https://link.springer.com/article/10.1007/s44372-025-00389-9]
- 83. Comparative analysis of ABCB1 reveals novel structural ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2014.00657/full]
- 84. Interactive effects of life cycle and monocot-dicot lineage ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1647198/full]
- 85. Interactive effects of life cycle and monocot-dicot lineage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12426603/]
- 86. 11 Annotation — Single- cell best practices [https://www.sc-best-practices.org/cellular_structure/annotation.html]
- 87. for scRNAseq - biostatsquid.com Cell type annotation [https://biostatsquid.com/cell-type-annotation-simply-explained/]
- 88. Automated cell from R to Loupe... | 10x Genomics type annotation [https://www.10xgenomics.com/analysis-guides/automated-cell-type-annotation-from-r-to-loupe-using-louper]