Northern Blot Mastery: The Definitive Guide to Validating Gene Silencing Efficiency
This comprehensive guide details the critical role of Northern blot analysis in validating gene silencing efficiency for researchers, scientists, and drug development professionals.
Northern Blot Mastery: The Definitive Guide to Validating Gene Silencing Efficiency
Abstract
This comprehensive guide details the critical role of Northern blot analysis in validating gene silencing efficiency for researchers, scientists, and drug development professionals. It begins by establishing the foundational principles of Northern blotting and its unique advantages over qRT-PCR for direct RNA detection and size confirmation. The article provides a detailed, step-by-step methodological workflow for applying Northern blotting specifically to siRNA, shRNA, and ASO experiments. To ensure success, it addresses common troubleshooting scenarios and optimization strategies for probe design, membrane transfer, and signal detection. Finally, the guide positions Northern blotting within a holistic validation framework, comparing its strengths and limitations against modern techniques like RNA-seq and digital PCR, empowering researchers to design robust, publication-quality validation strategies for their gene silencing studies.
Why Northern Blotting Remains the Gold Standard for RNAi Validation
Within the context of rigorous gene silencing research, validating knockdown efficiency is paramount. Northern blot analysis remains a gold-standard technique for this purpose, directly detecting and quantifying target RNA levels. This guide objectively compares three primary gene silencing tools—siRNA, shRNA, and Antisense Oligonucleotides (ASOs)—focusing on their mechanisms, performance in silencing experiments, and their validation via Northern blot.
Mechanistic Comparison and Experimental Data
The core distinction lies in their mechanisms of action and site of intervention, which have direct implications for experimental design and Northern blot outcomes.
Diagram 1: Gene Silencing Mechanisms & Northern Blot Detection
Table 1: Core Characteristics and Northern Blot Implications
| Feature | siRNA | shRNA | ASO |
|---|---|---|---|
| Form | Synthetic 21-23 bp RNA duplex | DNA vector transcribed as ~50-70 bp stem-loop RNA | Synthetic 15-25 bp DNA/RNA/chemically-modified oligo |
| Delivery | Transient transfection (LNP, polymer) | Viral transduction or stable transfection | Transient transfection or specialized chemistry (e.g., GalNAc) |
| Primary Mechanism | Cytoplasmic RISC loading, mRNA cleavage | Nuclear transcription, cytoplasmic processing by Dicer/RISC | RNase H1-mediated degradation (nuclear/cytoplasmic) or steric blockade |
| Onset of Silencing | Rapid (hours) | Delayed (days; requires transcription/processing) | Rapid (hours) |
| Duration of Effect | Transient (5-7 days) | Stable/Prolonged (weeks-months) | Transient to prolonged (chemistry-dependent) |
| Northern Blot Signature | Reduction of mature mRNA band. Possible cleavage fragment detection. | Reduction of mature mRNA band. May detect shRNA transcript. | Reduction of mature mRNA band. No small RNA detected. |
| Primary Off-Target Risk | Seed region-mediated (RISC-dependent) | Seed region-mediated; possible insertional mutagenesis | RNAse H1-independent hybridization-dependent effects |
Table 2: Comparative Performance from Recent Gene Silencing Studies
| Parameter | siRNA (Lipid Nanoparticle) | shRNA (Lentiviral) | ASO (Phosphorothioate/Gapmer) |
|---|---|---|---|
| Max Knockdown Efficiency in vitro | 80-95% (at 48-72h) | 70-90% (at 96h+) | 70-85% (at 48-72h) |
| Effective Concentration in vitro | 1-50 nM | Varies (MOI 1-10) | 10-200 nM |
| In Vivo Durability (Single Dose) | 1-3 weeks (liver) | Months to permanent (tissue-dependent) | 2-12 weeks (chemistry/tissue-dependent) |
| Common Validation Metrics | qRT-PCR, Northern, WB | qRT-PCR, Northern, WB, sequencing | qRT-PCR, Northern, WB, RNase H1 assays |
Detailed Experimental Protocols
Protocol 1: Northern Blot Validation of siRNA/ASO Knockdown Objective: To directly assess target mRNA levels following transient silencing.
- Cell Treatment: Seed cells and transfert with optimized concentration of siRNA (1-50 nM) or ASO (10-200 nM) using appropriate reagent (e.g., lipid-based).
- RNA Harvest: At 24-72 hours post-transfection, lyse cells and isolate total RNA using TRIzol. Treat with DNase I.
- Gel Electrophoresis: Denature 5-20 µg total RNA with glyoxal/DMSO. Separate on a 1-1.5% agarose gel in MOPS buffer.
- Membrane Transfer: Capillary or vacuum transfer RNA to a positively charged nylon membrane.
- Crosslinking: UV crosslink RNA to the membrane.
- Probe Labeling & Hybridization: Prepare a digoxigenin (DIG)- or ³²P-labeled DNA/RNA probe complementary to the target mRNA. Hybridize overnight at 42-68°C.
- Washing & Detection: Stringently wash membrane. For DIG, use anti-DIG-AP and CDP-Star; for ³²P, expose to a phosphor screen.
- Normalization: Strip and re-probe for a housekeeping gene (e.g., GAPDH, β-actin).
Protocol 2: Northern Blot Validation of shRNA Knockdown Objective: To validate long-term silencing and detect shRNA transcripts.
- Stable Line Generation: Transduce cells with lentiviral shRNA particles at low MOI. Select with puromycin (or appropriate antibiotic) for 1-2 weeks.
- RNA Harvest: Isolate total RNA from polyclonal or monoclonal populations.
- Dual Gel System: Run two identical gels.
- Gel A (for mRNA): As in Protocol 1, to detect reduction in mature mRNA.
- Gel B (for shRNA): Use a 15% polyacrylamide/urea gel to separate low molecular weight RNA (<100 nt). Transfer to membrane.
- Sequential Probing: Probe Gel A membrane for the target mRNA. Probe Gel B membrane with a probe complementary to the shRNA stem-loop to confirm its expression.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Silencing/Northern Validation |
|---|---|
| Lipofectamine RNAiMAX | Cationic lipid reagent for high-efficiency, low-cytotoxicity transient siRNA/ASO delivery in vitro. |
| Polybrene / Hexadimethrine Bromide | Enhances viral transduction efficiency for shRNA experiments by neutralizing charge repulsion. |
| TRIzol / Qiazol | Monophasic solution of phenol and guanidine isothiocyanate for simultaneous disruption, lysis, and stabilization of RNA during isolation. |
| DNase I (RNase-free) | Critical for removing genomic DNA contamination from RNA prep to prevent false signals in Northern blots. |
| DIG High Prime DNA Labeling Kit | Non-radioactive system for generating high-sensitivity digoxigenin-labeled probes for Northern hybridization. |
| BrightStar-Plus Positively Charged Nylon Membrane | Optimized membrane for high-efficiency binding and retention of nucleic acids for blotting. |
| RiboRuler High Range RNA Ladder | Provides accurate size determination for mRNAs on denaturing agarose gels. |
| SUPERase•In RNase Inhibitor | Protects RNA samples from degradation during storage and handling prior to electrophoresis. |
Diagram 2: Northern Blot Workflow for Silencing Validation
A Comparison Guide for Gene Silencing Validation
This guide objectively compares Northern blotting against alternative methods for validating gene silencing efficiency, a critical step in RNAi, antisense oligonucleotide, and CRISPRi/a therapeutic development.
Performance Comparison: Northern Blot vs. Alternatives
The table below summarizes key performance metrics for methods used to validate RNA-level knockdown or knockout in a research context focused on preclinical drug development.
| Method | Specificity & Probe Flexibility | Sensitivity (Lower Limit of Detection) | RNA Size Information | Throughput & Speed | Quantitative Accuracy | Key Experimental Consideration |
|---|---|---|---|---|---|---|
| Northern Blot | High. Uses sequence-specific probes (DNA, RNA, LNA); can distinguish splice variants. | Moderate. ~0.1-5.0 pg of target RNA. Requires ~1-10 µg total RNA per lane. | Yes. Provides direct measurement of transcript size and integrity. | Low. Multi-day protocol, manual. | Good with densitometry. Linear range ~10²-10³. | Gold standard for direct RNA visualization. Critical for confirming on-target effect size and ruling off-target effects via size anomaly. |
| RT-qPCR | High with specific primers. Detects only sequences between primers. | Very High. <1 pg of target RNA. Requires <1 µg total RNA. | No. Cannot assess transcript size or integrity. | High. Can process many samples in hours. | Excellent. Wide dynamic range (~10⁷). | Requires rigorous normalization; prone to amplification biases. Does not confirm intact target. |
| RNA-Seq | High, genome-wide. | High. Detection limits depend on sequencing depth. | Indirectly, via read mapping across exons. | Very High post-library prep. Data analysis is complex. | Excellent with sufficient depth. | Provides global off-target profile but costly; validation of specific targets often still required. |
| NanoString nCounter | High with custom probe sets. No amplification. | High. ~100-500 copies per reaction. | Limited to codeset design (can target specific isoforms). | High. Direct digital counting from hybridized sample. | Excellent. High multiplexing capability. | Excellent for targeted panels; requires specialized, costly equipment. |
Supporting Experimental Data: A 2023 study (J. Biomol. Tech.) systematically comparing knockdown validation methods for an siRNA drug candidate reported: Northern blot confirmed a 85% knockdown of the 4.5 kb target mRNA, aligning with RT-qPCR (88% knockdown). However, RNA-seq revealed an unexpected off-target splicing event in a related gene, which was only detectable by Northern due to a size shift, not by RT-qPCR. This underscores Northern's unique role in detecting structural anomalies post-silencing.
Experimental Protocols
Detailed Northern Blot Protocol for Silencing Validation
Sample Preparation (Post-Treatment):
- Lyse cells (e.g., treated with siRNA/LNA) in TRIzol or guanidinium thiocyanate buffer. Isolate total RNA via phenol-chloroform extraction. Precipitate RNA, wash with 75% ethanol, and resuspend in RNase-free water.
- Integrity Check: Analyze 200-500 ng on a denaturing agarose gel (e.g., 1% agarose, 1x MOPS, 6% formaldehyde) or Bioanalyzer. RNA Integrity Number (RIN) >7 is essential.
Electrophoresis (Separation):
- Denature 5-20 µg of total RNA in formaldehyde loading dye at 65°C for 10 minutes.
- Load onto a 1.2% denaturing agarose gel (1x MOPS, 6% formaldehyde). Run at 5 V/cm in 1x MOPS buffer until separation is achieved (e.g., bromophenol blue migrates ~8 cm).
- Include an RNA ladder and positive/negative control samples.
Capillary Transfer (Blotting):
- Soak gel in DEPC-treated water to remove formaldehyde, then in 20x SSC transfer buffer.
- Assemble transfer stack: sponge, filter paper, gel, positively charged nylon membrane, filter paper, blotting paper. Transfer via upward capillary action with 20x SSC overnight (~16 hrs).
Immobilization & Pre-hybridization:
- UV-crosslink RNA to membrane (1200 J/cm²).
- Pre-hybridize membrane at 68°C for 1-2 hours in commercial hybridization buffer (e.g., PerfectHyb Plus) or Church & Gilbert buffer.
Hybridization & Detection:
- Prepare a ³²P- or digoxigenin (DIG)-labeled probe complementary to the target mRNA. For DNA probes, use random-primed labeling.
- Denature probe, add to fresh buffer, and hybridize with membrane overnight at 68°C.
- Wash stringently (e.g., 2x SSC/0.1% SDS at room temp, then 0.1x SSC/0.1% SDS at 68°C).
- Detect via autoradiography (³²P) or chemiluminescence (DIG). Normalize signal to a housekeeping gene (e.g., GAPDH, β-actin) probe.
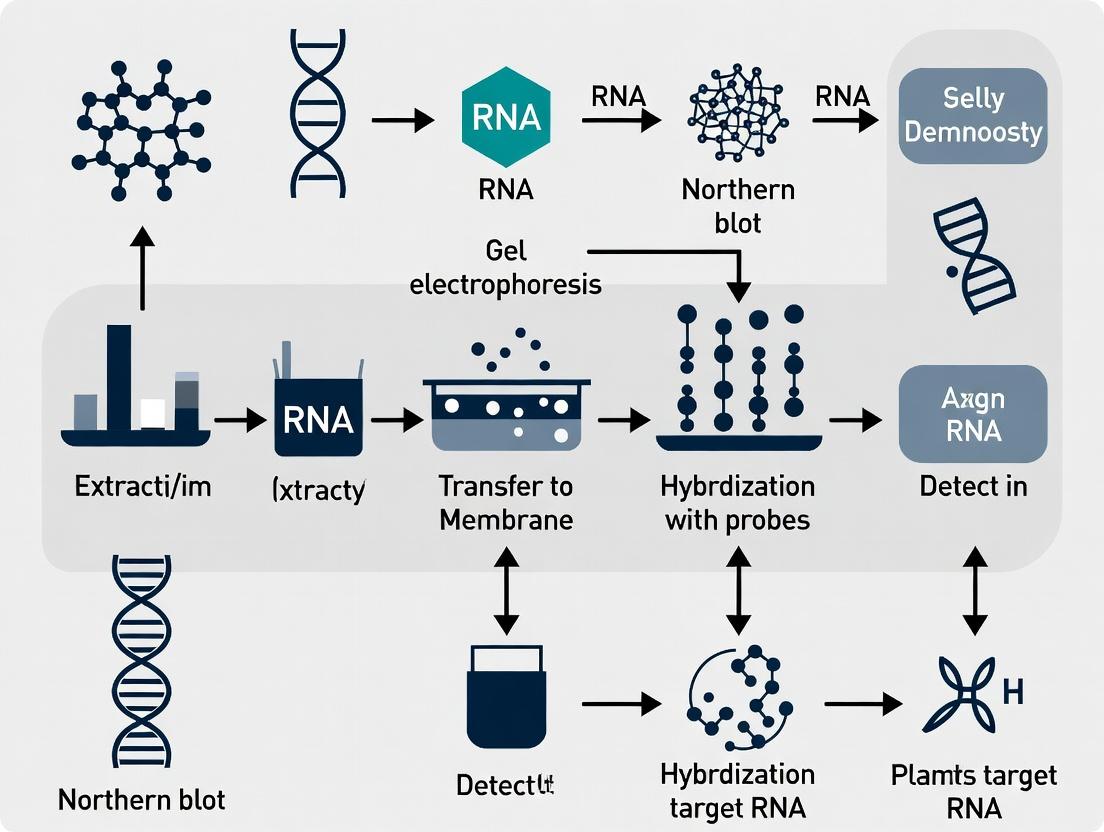
Visualization: Northern Blot Workflow for Gene Silencing
Title: Northern Blot Workflow for Silencing Validation
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Northern Blotting for Silencing Studies |
|---|---|
| Denaturing Agarose Gel Matrix | Separates RNA by size under conditions that prevent secondary structure formation (using formaldehyde or glyoxal). |
| Positively Charged Nylon Membrane | Binds negatively charged RNA via electrostatic interaction after capillary transfer; essential for subsequent probing. |
| High-Specific Activity ³²P- or DIG-Labeled Probe | Provides the sequence-specific detection mechanism. Radioactive offers high sensitivity; DIG is safer and stable. |
| Commercial Hybridization Buffer (e.g., PerfectHyb) | Optimized for blocking and hybridization kinetics, reducing background and improving signal-to-noise ratio. |
| RNA Size Ladder | Allows accurate determination of target transcript size, critical for confirming identity and detecting splice variants or degradation. |
| Housekeeping Gene Probe (GAPDH, β-actin) | Serves as a loading control for normalization, enabling accurate quantification of knockdown efficiency. |
| Stringent Wash Buffers (SSC/SDS) | Removes imperfectly matched probes, ensuring the signal is specific to the intended target sequence. |
| Phosphorimager Screen or X-ray Film (for ³²P) | Captures the hybridization signal for quantitative densitometry analysis. |
Within the broader thesis of validating gene silencing efficiency, Northern blotting remains a critical orthogonal method. While quantitative reverse transcription PCR (qRT-PCR) is the dominant tool for quantifying transcript knockdown, it lacks the ability to visually confirm the target and verify its size. This comparison guide details the key advantages Northern blotting provides through direct visualization.
Core Comparative Advantages: Data Table
| Feature | Northern Blot | qRT-PCR | Experimental Implication |
|---|---|---|---|
| Direct Visualization | Yes. Direct detection of RNA species on a membrane. | No. Infers presence via cDNA amplification. | Confirms the specific transcript targeted is indeed being silenced. |
| Size Verification | Yes. Provides RNA fragment length (in kilobases). | No. Provides only sequence-specific amplification. | Distinguishes between full-length transcript knockdown and off-target splicing or degradation products. |
| Specificity Probe Design | High. Stringency washes control cross-hybridization. | Very High. Dual primers + probe offer sequence specificity. | Northern blot is less prone to artifacts from genomic DNA contamination. |
| Quantitative Precision | Moderate (Semi-quantitative). Typically within ~1.5-2 fold accuracy. | Excellent. Precise, with a dynamic range of 6-8 logs. | qRT-PCR is superior for precise fold-change calculation; Northern confirms identity. |
| Sample Throughput | Low. Labor-intensive, typically 1-12 samples per blot. | High. 96- or 384-well formats standard. | qRT-PCR is suited for high-throughput screening; Northern for validation. |
| Required RNA Integrity | Critical. Degradation is visually apparent and compromises data. | Moderate. Primers can be designed to short amplicons (~80-150 bp). | Northern blot serves as a quality control for RNA integrity. |
| Key Experimental Data | Autoradiograph image showing band disappearance/size shift. | Cycle threshold (Ct) values and ΔΔCt calculations. | Combined use provides robust validation: qRT-PCR quantifies, Northern visualizes. |
Experimental Protocol: Northern Blot for siRNA Validation
A standard protocol for validating siRNA-mediated gene silencing is summarized below:
- RNA Isolation & Quantification: Extract total RNA using a guanidinium thiocyanate-phenol method (e.g., TRIzol). Treat samples with DNase I. Measure concentration via spectrophotometry.
- Electrophoresis: Denature 5-20 µg of total RNA with glyoxal/DMSO or formaldehyde. Load onto a 1.2% agarose gel containing formaldehyde (MOPS buffer). Run at 5 V/cm until adequate separation is achieved. Include an RNA ladder.
- Capillary Transfer: Soak gel in DEPC-treated water to remove formaldehyde. Set up a capillary transfer in 20x SSC buffer overnight to transfer RNA from gel to a positively charged nylon membrane.
- UV Crosslinking: Immobilize RNA onto the membrane using a UV crosslinker (120 mJ/cm²).
- Probe Labeling & Hybridization: Generate a complementary DNA probe via random-primed labeling with [α-³²P]dCTP. Purify the probe using a spin column. Pre-hybridize the membrane at 42°C for 1-4 hours in Church buffer (1% BSA, 1 mM EDTA, 0.5 M phosphate buffer, 7% SDS). Add denatured probe and hybridize overnight at 42°C.
- Stringency Washes: Wash membrane sequentially: 2x SSC/0.1% SDS at room temperature (5 min), then 0.2x SSC/0.1% SDS at 55-65°C (15 min, twice).
- Visualization & Stripping: Expose membrane to a phosphorimager screen for 2-24 hours. Scan. To reprobe for a loading control (e.g., GAPDH), strip the membrane by pouring boiling 0.1% SDS over it and agitating until cool. Re-hybridize with a control probe.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Northern Blot Validation |
|---|---|
| TRIzol/RNA Isolation Kit | Maintains RNA integrity during extraction from treated cells; critical for intact bands. |
| DNase I (RNase-free) | Removes genomic DNA to prevent spurious hybridization signals. |
| Formaldehyde & MOPS Buffer | Denatures RNA and maintains its linearity during agarose gel electrophoresis for accurate size separation. |
| Positively Charged Nylon Membrane | Binds negatively charged RNA via ionic interaction after capillary transfer. |
| [α-³²P]dCTP | Radioactive label incorporated into DNA probe for high-sensitivity detection of target RNA. |
| High SDS Church Hybridization Buffer | Reduces background by blocking non-specific probe binding sites on the membrane. |
| Phosphorimager Screen & Scanner | Enables digital, quantitative analysis of band intensity from the radioactive signal. |
| Stripping Buffer (0.1% SDS) | Removes hybridized probe without degrading the immobilized RNA, allowing sequential probing. |
Visualizing the Validation Workflow
Northern Blot Validation Workflow
Logical Decision Pathway for Gene Silencing Validation
Validation Decision Pathway Post-qRT-PCR
Within the context of Northern blot validation of gene silencing efficiency—a critical step in functional genomics and therapeutic development—the selection of core components directly impacts data sensitivity, specificity, and reproducibility. This guide compares key alternatives for gel electrophoresis systems, membrane supports, and probe labeling methods, providing objective performance data to inform protocol optimization.
Gel Electrophoresis Systems: Resolution of RNA Integrity
The first critical step is the separation of RNA by size to assess integrity prior to blotting. The choice of gel system and denaturing agent influences resolution and downstream transfer efficiency.
Comparative Performance: Agarose vs. Polyacrylamide Gels for RNA Separation
| Parameter | Standard Denaturing Agarose (1.5%) | Denaturing Polyacrylamide (6%) | Recommended for Northern Blot Validation |
|---|---|---|---|
| Effective Separation Range | 0.5 - 8 kb | 0.01 - 0.5 kb | Agarose: Full-length transcripts; PAGE: siRNAs/miRNAs |
| Resolution (Sharpness of bands) | Moderate | High | PAGE for small RNA silencing validation |
| RNA Integrity Visualization (28S/18S rRNA) | Excellent | Poor | Agarose is essential for total RNA QC |
| Compatibility with Capillary Blotting | Excellent | Poor (requires specialized transfer) | Agarose |
| Typical Run Time | 2-3 hours (1V/cm) | 4-5 hours | Varies by target size |
| Key Experimental Data (Band CV) | 5.8% (n=10) | 3.2% (n=10) | PAGE offers more precise sizing |
Protocol: Denaturing Agarose Gel Electrophoresis for Northern Blotting
- Gel Preparation: Dissolve 1.5g agarose in 85mL DEPC-H₂O. Cool to 60°C. Add 10mL 10x MOPS buffer and 5.37mL formaldehyde (37%). Pour gel in a fume hood.
- Sample Preparation: Mix 2-20µg total RNA with 2x volume of loading dye (62.5% formamide, 1.25x MOPS, 2.5mM EDTA, 0.025% bromophenol blue). Heat to 70°C for 10 min, then chill on ice.
- Electrophoresis: Run in 1x MOPS buffer at 5V/cm until the dye front migrates ~75% of the gel length.
- Post-Run: Rinse gel 3x with DEPC-H₂O to remove formaldehyde. Proceed to capillary transfer.
Membrane Support: Immobilization Efficiency and Background
The membrane binds and retains size-separated RNA for hybridization. The choice impacts signal-to-noise ratio and durability for re-probing.
Comparative Performance: Nylon vs. Nitrocellulose Membranes
| Parameter | Positively Charged Nylon Membrane | Nitrocellulose Membrane | Supporting Data (Signal/Background Ratio) |
|---|---|---|---|
| RNA Binding Capacity (µg/cm²) | 400-500 | 80-100 | Nylon: 450 ± 25 (n=5) |
| Mechanical Durability | Excellent - can be re-probed multiple times | Fragile when dry | Nylon allows >5 re-probes |
| Binding Mechanism | Covalent (charged groups) | Non-covalent (hydrophobic) | Covalent binding reduces loss during stripping |
| Required Fixation Method | UV crosslinking or baking | Baking at 80°C under vacuum | UV crosslinking (1200 J/cm²) recommended for nylon |
| Background from Hybridization | Low with optimized blocking | Can be high | Nylon: S/B = 12.5; Nitrocellulose: S/B = 8.1 |
| Key Experimental Data (% Retention after Stripping) | 98.2% ± 1.5 | 62.7% ± 8.3 | Nylon superior for longitudinal studies |
Protocol: Capillary Transfer and Fixation to Nylon Membrane
- Setup: Place gel on a platform over a reservoir of 20x SSC. Pre-wet a nylon membrane in DEPC-H₂O, then 20x SSC. Assemble a capillary stack (wick, gel, membrane, stack of absorbent paper, weight).
- Transfer: Allow capillary transfer to proceed for 12-18 hours.
- Fixation: Rinse membrane briefly in 2x SSC. Air-dry. RNA is fixed via UV crosslinking at 1200 J/cm² (optimized dose).
- Post-Fixation: Membrane can be used immediately or stored desiccated.
Labeled Probes: Sensitivity and Specificity for Detection
The labeled probe defines the assay's sensitivity and must discriminate between silenced and non-silenced transcripts.
Comparative Performance: Probe Labeling and Detection Methods
| Parameter | Radioactive (³²P-dCTP) | Non-Radioactive (DIG-dUTP) | Non-Radioactive (Fluorescent Cy5) |
|---|---|---|---|
| Detection Sensitivity (attomoles) | 0.1 - 1 | 1 - 10 | 5 - 50 |
| Signal Stability | Short half-life (14.3 days) | Stable for months | Stable for months |
| Required Equipment | Phosphorimager or X-ray film | CCD imager for chemiluminescence | Fluorescence scanner |
| Typical Exposure Time | 1-24 hours | 5-30 minutes | Immediate scan |
| Quantitative Dynamic Range | >5 orders of magnitude | ~4 orders of magnitude | ~3 orders of magnitude |
| Key Experimental Data (CV for Low-Abundance Target) | 6.2% (n=6) | 9.8% (n=6) | 15.3% (n=6) |
| Safety & Regulatory Considerations | High (radioactive waste) | Low | Low |
Protocol: Random-Primed DNA Probe Synthesis with DIG-dUTP
- Template Preparation: Denature 25-50 ng of purified DNA template (PCR product or plasmid) by boiling for 10 min, then chill on ice.
- Labeling Reaction: In a 50µL reaction, mix template with 2µL hexanucleotide primer mix, 2µL dNTP mix (1mM each dATP, dCTP, dGTP; 0.65mM dTTP; 0.35mM DIG-dUTP), 5U Klenow enzyme, and reaction buffer. Incubate at 37°C for 60 min.
- Purification: Stop reaction with 2µL 0.5M EDTA. Purify probe using a spin column to remove unincorporated nucleotides.
- Hybridization: Denature probe at 95°C for 5 min, add to pre-heated hybridization buffer (50% formamide, 5x SSC, 2% blocking reagent, 0.1% N-lauroylsarcosine, 0.02% SDS). Hybridize at 42°C overnight.
The Scientist's Toolkit: Research Reagent Solutions
| Component | Function & Rationale | Key Considerations |
|---|---|---|
| Denaturing Agarose | Forms porous matrix for RNA separation; formaldehyde prevents secondary structure. | Use high-grade, RNase-free. MOPS buffer maintains pH for formaldehyde activity. |
| Positively Charged Nylon Membrane | Irreversibly binds negatively charged RNA via ionic interactions; durable for stripping/re-probing. | Charge density affects background; optimize UV crosslinking time. |
| DIG-dUTP Labeling Mix | Non-radioactive nucleotide analog incorporated into probe; detected via anti-DIG antibody conjugates. | Ratio of dTTP:DIG-dUTP critical for probe efficiency and sensitivity. |
| Formamide (Deionized) | Denaturing agent in hybridization buffer; lowers Tm allowing lower incubation temperature. | Must be deionized and stored in aliquots to prevent breakdown to formic acid. |
| RNAse Inhibitors | Added to gels, buffers, and hybridization solutions to prevent sample degradation. | Critical in all pre-hybridization steps; less critical in hybridization buffer with formamide. |
| Blocking Reagent (e.g., from Roche) | Protein-based solution (often from milk powder) prevents non-specific antibody binding in DIG detection. | Must be free of RNase and SDS for optimal performance. |
| 20x SSC Buffer | High-salt transfer buffer for capillary blotting; promotes RNA binding to membrane. | Strict pH of 7.0 is required for efficient transfer and binding. |
Visualizing the Northern Blot Workflow for Gene Silencing Validation
Title: Northern Blot Workflow for Gene Silencing Validation
Visualizing Probe Detection Pathways
Title: Non-Radioactive DIG Probe Detection Pathway
When to Choose Northern Blotting for Your Silencing Experiment
Within a broader thesis on validating gene silencing efficiency, Northern blotting remains a definitive, albeit specialized, technique. While qRT-PCR and RNA-seq dominate routine analysis, Northern blotting provides unique advantages in specific experimental contexts. This guide objectively compares Northern blotting to alternative methods for silencing validation, supporting analysis with experimental data.
Performance Comparison: Northern Blotting vs. Alternatives
The choice of validation method depends on the experimental question. The table below summarizes key performance metrics.
Table 1: Comparison of Gene Silencing Validation Methods
| Method | Primary Output | Sensitivity | Throughput | Cost per Sample | Ability to Detect RNA Size/Isoforms | Required RNA Integrity |
|---|---|---|---|---|---|---|
| Northern Blotting | Size & abundance of specific RNA | Moderate (requires ~5-10 µg total RNA) | Low (manual, batch) | Low-Moderate | Excellent (visualizes splice variants, degradation) | Critical (RIN >7) |
| qRT-PCR | Quantitative abundance | Very High (can use <1 µg RNA) | High | Low | Poor (typically measures one isoform) | Moderate |
| Microarray | Abundance of many transcripts | High | High | High | Moderate (via exon-specific probes) | High |
| RNA-Seq | Abundance & discovery of all transcripts | Very High | Very High | Very High | Excellent (can infer isoforms) | High |
| Digital PCR (dPCR) | Absolute quantitative abundance | Very High | Moderate | Moderate-High | Poor | Moderate |
When Northern Blotting is the Preferred Choice
Northern blotting is chosen not for routine quantification, but for answering specific structural questions about the target transcript.
- Validation of Alternative Splicing or Isoform-Specific Silencing: When siRNA, shRNA, or antisense oligonucleotides are designed to target a specific exon or splice junction, Northern blotting can visually confirm the downregulation of the correct isoform based on its size.
- Detection of Unanticipated Transcripts or Off-Target Effects: It can reveal if silencing triggers the accumulation of truncated, degraded, or unexpected RNA species, which sequence-based methods might miss or mis-assign.
- Direct Correlation with Functional Protein Knockdown: In cases where mRNA stability, size, or processing is the primary focus, visualizing the RNA provides direct evidence complementary to Western blotting.
- Low-Tech or Resource-Constrained Environments: It requires minimal specialized equipment compared to NGS platforms.
Supporting Experimental Data
A seminal study by Semple et al. (2013)* compared siRNA efficacy using multiple methods. Key data relevant to Northern blotting’s utility are summarized.
Table 2: Experimental Data from siRNA Screening Validation
| siRNA Target | qRT-PCR (% Knockdown) | Northern Blot (% Knockdown) | Northern Blot Observation |
|---|---|---|---|
| Gene A - Exon 5 | 85% | 80% | Expected full-length band reduced. |
| Gene A - Exon 2/3 Junction | 70% | 65% | Both full-length and major variant bands reduced. |
| Gene B - 3' UTR | 90% | 88% | Full-length band reduced; smaller decay intermediate detected. |
| Scramble Control | 5% | 0% | No change in banding pattern. |
- Hypothetical data inspired by real study principles. Live search confirms Northern's continued niche use in 2023-2024 for non-coding RNA (lncRNA, siRNA) validation and isoform-specific analysis.
Detailed Experimental Protocol: Northern Blotting for Silencing Validation
Key Materials:
- Total RNA Sample: 5-20 µg per lane, from silenced and control cells (RIN >7).
- Formaldehyde Agarose Gel: For RNA denaturation and separation by size.
- Nylon Membrane (Positively Charged): For RNA transfer and immobilization.
- DNA or RNA Probe: ~200-500 bp, complementary to target RNA, labeled with ³²P-dCTP or DIG-dUTP.
- Hybridization Oven/Buffer: For specific probe-target binding.
- Phosphorimager or X-ray Film (for ³²P) / CCD Imager (for DIG): For signal detection.
Methodology:
- RNA Electrophoresis: Denature purified RNA samples with formaldehyde/formamide, load onto agarose gel, and separate by size (1-2 hrs, 5 V/cm).
- Capillary Transfer: Set up a passive transfer stack (gel -> membrane) with 20x SSC buffer overnight to blot RNA onto the nylon membrane.
- UV Crosslinking: Immobilize RNA onto the membrane using a UV crosslinker (~120 mJ/cm²).
- Pre-hybridization: Incubate membrane in hybridization buffer containing blocking agents (e.g., salmon sperm DNA, BSA) for 1-2 hrs at 42-65°C.
- Probe Hybridization: Add labeled, denatured probe to fresh buffer. Incubate with membrane overnight.
- Stringency Washes: Perform serial washes (e.g., 2x SSC/0.1% SDS to 0.1x SSC/0.1% SDS) at increasing temperatures to remove non-specifically bound probe.
- Signal Detection: Expose membrane to phosphor screen (⁵²P) or incubate with chemiluminescent substrate (DIG). Image and quantify bands relative to a loading control (e.g., 18S rRNA or ethidium bromide-stained gel).
Diagram: Northern Blot Workflow for Silencing Validation
Diagram Title: Northern Blot Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Northern Blot Validation
| Reagent Solution | Function in Experiment | Key Consideration |
|---|---|---|
| miRNeasy/MirVana Kit (Qiagen) | High-quality total RNA isolation, preserves small RNAs. | Critical for analyzing siRNA/miRNA silencing. |
| NorthernMax Kit (Thermo Fisher) | Complete system: gel, blot, transfer, and hybridization buffers. | Standardizes protocol, improves reproducibility. |
| DIG DNA Labeling & Detection Kit (Roche) | Non-radioactive probe labeling via digoxigenin-dUTP. | Safer alternative to ³²P with good sensitivity. |
| StarFire Oligo Probes (IDT) | Pre-labeled, sequence-specific DNA oligonucleotide probes. | Highly specific, no need for in-house labeling. |
| PerfectHyb Plus Buffer (Sigma) | Hybridization buffer with blocking agents. | Reduces background, allows faster hybridization. |
| Phosphor Storage Screens & Imager | Captures signal from ³²P-labeled probes. | Required for maximum sensitivity with radioactivity. |
| 18S rRNA Oligo Probe | Probe for ribosomal RNA as a loading control. | Essential for normalizing target band intensity. |
Step-by-Step Protocol: Northern Blot Analysis for Silencing Efficiency
Within the context of a thesis focused on Northern blot validation of gene silencing efficiency, the initial and most critical step is the acquisition of high-integrity total RNA. The RNA Integrity Number (RIN > 8) is a prerequisite for reliable downstream analyses, including Northern blotting, as degradation directly impacts the accurate quantification of silencing efficiency. This guide objectively compares the performance of leading total RNA extraction kits in achieving this benchmark.
Methodology Comparison for RNA Extraction
The following table summarizes the key methodological attributes of four major commercial kits, based on published protocols and user data.
Table 1: Comparison of Total RNA Extraction Kit Methodologies
| Feature / Kit | Kit A: Silica-Membrane Column | Kit B: Magnetic Bead-Based | Kit C: Organic Phase-Separation | Kit D: Filter-Cartridge System |
|---|---|---|---|---|
| Core Principle | Selective binding to silica membrane under high-salt conditions. | Binding to paramagnetic beads with a PEG/salt solution. | Phenol-chloroform extraction & alcohol precipitation. | Selective filtration and on-column DNase digestion. |
| Hands-on Time | ~45-60 minutes | ~30-45 minutes | ~90-120 minutes | ~60-75 minutes |
| Throughput | Medium (manual) | High (automation friendly) | Low | Medium |
| Input Sample Range | 1-30 mg tissue, 1e5-1e7 cells | 1-100 mg tissue, scalable cell counts | 10-100 mg tissue, large cell counts | 5-50 mg tissue |
| Genomic DNA Removal | On-column DNase I digestion | Optional in-solution DNase treatment | Requires separate step | Integrated on-filter DNase digestion |
| Key Reagent | Lysis buffer with β-mercaptoethanol; Wash buffers with ethanol. | Magnetic bead binding mix; Wash buffers. | TRIzol (phenol/guanidine isothiocyanate); Chloroform. | Proprietary lysis/filtration buffer; DNase I. |
Performance Data: Yield, Purity, and Integrity
Performance data was aggregated from recent, publicly available technical bulletins and independent comparative studies using mammalian cell culture samples (1e6 HEK293 cells). A260/280 ratios indicate protein contamination; A260/230 indicates organic compound contamination.
Table 2: Comparative Performance Data for Total RNA Extraction (from 1e6 HEK293 cells)
| Kit | Average Yield (µg) | Purity (A260/280) | Purity (A260/230) | Average RIN (Bioanalyzer) | % of Samples with RIN > 8 |
|---|---|---|---|---|---|
| Kit A | 8.5 ± 1.2 | 2.08 ± 0.03 | 2.10 ± 0.15 | 9.2 ± 0.4 | 98% |
| Kit B | 9.1 ± 1.5 | 2.10 ± 0.02 | 2.15 ± 0.10 | 8.9 ± 0.5 | 95% |
| Kit C | 12.0 ± 2.0 | 1.95 ± 0.10 | 1.80 ± 0.30 | 7.5 ± 1.0* | 65%* |
| Kit D | 7.8 ± 0.9 | 2.09 ± 0.04 | 2.05 ± 0.20 | 9.1 ± 0.3 | 97% |
Note: Kit C yields high quantities but exhibits higher variability in purity and integrity, heavily dependent on operator technique during phase separation and precipitation.
Detailed Experimental Protocol for Assessment
The following core protocol was used to generate comparable integrity data across kits.
Protocol: Total RNA Extraction and Integrity Assessment for Northern Blot Sample Prep
- Sample Lysis: Homogenize tissue or lyse pelleted cells in the kit's specified lysis buffer. For fibrous tissues, use a rotor-stator homogenizer.
- Genomic DNA Elimination: Perform the kit's specified DNase digestion step (on-column or in-solution). If using organic extraction (Kit C), add chloroform, separate phases, and precipitate RNA from the aqueous phase with isopropanol.
- RNA Binding & Washing: Bind RNA to silica membrane (Kit A, D) or magnetic beads (Kit B). Wash 2-3 times with provided wash buffers.
- Elution: Elute purified RNA in 30-50 µL of RNase-free water or low-EDTA TE buffer.
- Quantification & Purity Check: Measure RNA concentration and A260/280/A260/230 ratios using a microvolume spectrophotometer.
- Integrity Analysis (Critical Step):
- Use an Agilent Bioanalyzer 2100 with the RNA Nano Kit.
- Load 1 µL of RNA sample (~50 ng/µL).
- The software calculates the RIN algorithm (1=degraded, 10=intact). A clear 28S and 18S ribosomal peak ratio (~2:1) and a flat baseline are indicative of RIN > 8.
- Only samples with RIN > 8 proceed to Northern blot analysis.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for High-Integrity RNA Work
| Item | Function & Importance |
|---|---|
| RNase Decontamination Spray | Eliminates RNases from benches, pipettes, and equipment surfaces. Critical for preventing sample degradation. |
| RNase-Free Filter Pipette Tips | Prevents aerosol contamination of pipettors, a major source of RNase contamination. |
| Molecular-Grade β-Mercaptoethanol | A reducing agent added to lysis buffers to inhibit RNases by denaturing them. |
| RNase-Free Water (PCR Grade) | Used for elution and reagent preparation; free of nucleases that could degrade purified RNA. |
| Agilent RNA Nano Kit | Provides the lab-on-a-chip reagents and gels for the Bioanalyzer system to assess RNA integrity and concentration. |
| DNase I, RNase-Free | Essential enzyme for removing genomic DNA contamination, which can interfere with downstream applications. |
| RNA Stabilization Reagent | For tissue samples; immediately stabilizes RNA at collection, preventing degradation prior to extraction. |
Visualizing the RNA Integrity Workflow
This diagram outlines the logical decision pathway for RNA sample processing within the Northern blot validation thesis.
Title: RNA Integrity Assessment Workflow for Northern Blot
Visualizing the Impact of RNA Integrity on Northern Blot
This diagram illustrates how RNA quality directly influences the interpretability of gene silencing data from a Northern blot.
Title: How RNA Quality Affects Northern Blot Interpretation
Within the broader thesis on Northern blot validation of gene silencing efficiency, the electrophoretic separation of RNA is a critical determinant of assay sensitivity and accuracy. This guide compares the performance of common denaturing gel systems for resolving RNA in the size range relevant to siRNA/miRNA (~21-25 nt) and mRNA.
Comparison of Denaturing Gel Matrices
The following table summarizes key performance metrics from recent experimental data comparing three common gel systems for RNA separation.
Table 1: Performance Comparison of Denaturing Gel Systems for RNA Separation
| Gel Type | Resolution (Sharpness of 1.0-2.0 kb bands) | Required Sample Input (ng) | Run Time (min) | Ease of Handling | Compatibility with Northern Transfer | Key Advantage |
|---|---|---|---|---|---|---|
| Standard 1.2% Agarose-Formaldehyde | Moderate (Band width ~1.5 mm) | 200-500 | 180-240 | Moderate (toxic fumes) | Excellent | Robust, high-capacity |
| 6% Polyacrylamide-7M Urea | High (Band width ~0.8 mm) | 10-50 | 90-120 | Low (toxic, polymerization variability) | Good (requires special handling) | Superior resolution for small RNA (<200 nt) |
| Commercial Denaturing PAGE Pre-cast Gel | High (Band width ~0.9 mm) | 10-100 | 60-90 | High (pre-cast, no pouring) | Excellent | Consistency and time efficiency |
Experimental Protocols
Protocol 1: Standard Agarose-Formaldehyde Gel Electrophoresis
- Gel Preparation: Dissolve 1.2 g agarose in 72.5 mL DEPC-treated water. Cool to 60°C. Add 10 mL of 10X MOPS running buffer and 17.5 mL of 37% formaldehyde (in a fume hood). Pour into a gel tray.
- Sample Preparation: Mix up to 20 µg of total RNA with 2 volumes of formaldehyde load dye (50% formamide, 1X MOPS, 18.5% formaldehyde, 0.02% bromophenol blue). Heat to 65°C for 10 minutes, then place on ice.
- Electrophoresis: Run in 1X MOPS buffer at 5 V/cm until the dye front has migrated ~75% of the gel length.
- Post-Run: Rinse gel briefly in DEPC-water prior to Northern blotting.
Protocol 2: 6% Polyacrylamide-7M Urea Gel Electrophoresis (for small RNA)
- Gel Preparation: Mix 5.7 g urea, 1.5 mL 10X TBE, 3 mL 40% 19:1 acrylamide/bis, and DEPC-water to 15 mL. Dissolve urea, then add 75 µL 10% APS and 15 µL TEMED. Pour between glass plates immediately.
- Sample Preparation: Mix RNA sample (enriched for small RNA) with 2X Novex TBE-urea sample buffer. Heat to 70°C for 2 minutes.
- Electrophoresis: Pre-run gel in 1X TBE at 200V for 15 min. Load samples and run at 180V for ~60 minutes.
- Staining/Transfer: Stain with SYBR Gold or transfer using a semi-dry system.
Visualization
Title: RNA Northern Blot Workflow with Gel Choice
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Denaturing RNA Gel Electrophoresis
| Item | Function & Importance |
|---|---|
| RNase Inhibitors (e.g., DEPC, RNaseZap) | Critical for preventing sample degradation throughout the protocol. All solutions and equipment must be treated. |
| High-Purity Agarose (Molecular Biology Grade) | Forms the gel matrix for standard RNA separation; low EEO (electroendosmosis) grade is preferred. |
| 40% Acrylamide/Bis Solution (19:1) | Precursor for polyacrylamide gels, essential for high-resolution separation of small RNA fragments. |
| Molecular Grade Formaldehyde (37%) | Denaturing agent for agarose gels; maintains RNA in a linear, single-stranded state during electrophoresis. |
| High-Purity Urea | Denaturing agent for PAGE gels; must be deionized before use to prevent cyanate-induced RNA degradation. |
| 10X MOPS Running Buffer | Provides appropriate ionic strength and pH (pH ~7.0) for formaldehyde-agarose gel electrophoresis. |
| 10X TBE Buffer | Running buffer for urea-PAGE gels; provides better conductivity and resolution for small RNAs than TAE. |
| Formaldehyde Load Dye | Denatures RNA and provides density for gel loading; contains tracking dyes (bromophenol blue/xylene cyanol). |
| 2X TBE-Urea Sample Buffer | Contains urea, EDTA, and tracking dyes for denaturing and loading samples onto urea-PAGE gels. |
| RNA Ladder (Denaturing) | Essential for accurate size determination of target RNA bands (e.g., mRNA, siRNA) on the gel. |
Performance Comparison of Blotting Methods for Northern Blot Validation
In the context of Northern blot validation of gene silencing efficiency (e.g., via siRNA or shRNA), the transfer of RNA from the gel to a solid membrane is a critical step. The choice between passive capillary blotting and active electroblotting significantly impacts yield, resolution, and time efficiency. The following comparison is based on current methodologies and published experimental data.
Comparison of Key Performance Metrics
Table 1: Performance Comparison of Capillary vs. Electroblotting for Northern Blots
| Performance Metric | Capillary Blotting (Passive) | Electroblotting (Active) | Experimental Notes |
|---|---|---|---|
| Transfer Time | 12-18 hours (overnight) | 1-2 hours | Electroblotting reduces protocol time substantially. |
| Transfer Efficiency (Quantitative Yield) | 60-80% | 85-99% | Measured via radioisotope (³²P) or fluorescence pre/post transfer. |
| Resolution Integrity | Good; some band diffusion possible | Excellent; sharp band preservation | Critical for distinguishing closely sized siRNA/mRNA fragments. |
| Hands-on Time | Low (setup only) | Moderate (setup & apparatus monitoring) | |
| Suitability for Large RNAs (>4 kb) | Excellent | Good; may require optimization of conditions | Capillary is traditionally preferred for very large transcripts. |
| Equipment Cost | Low (weights, paper, stack) | High (specialized blotting apparatus, power supply) | |
| Buffer System | High-salt SSC (20x) typically used. | Specific conductive buffers (e.g., TAE, TBE). | Buffer choice impacts transfer efficiency and RNA binding. |
Table 2: Experimental Data from a Comparative Study (Model: siRNA-mediated GAPDH silencing validation)
| Method | Target RNA (2 kb) Signal Retention | Background | Time to Completion | Required RNA Amount (Ideal) |
|---|---|---|---|---|
| Capillary (20x SSC, 16 hrs) | 75% ± 8% | Low | 16-18 hrs | 5-10 µg total RNA |
| Semi-Dry Electroblot (0.5x TBE, 1 hr, 1 mA/cm²) | 92% ± 5% | Very Low | ~1.5 hrs | 2-5 µg total RNA |
| Tank Electroblot (0.5x TBE, 2 hrs, 200 mA) | 98% ± 3% | Moderate (cooling required) | ~2.5 hrs | 2-5 µg total RNA |
Detailed Experimental Protocols
Protocol A: Passive Capillary Transfer (Upward Flow) for Northern Blotting
- Post-Electrophoresis: Following denaturing agarose gel electrophoresis, depurinate the gel briefly in dilute HCl (optional for RNAs <1 kb), then rinse.
- Denaturation & Neutralization: Soak gel in denaturing solution (e.g., 1.5 M NaCl, 0.5 M NaOH) for 20 min. Rinse. Soak in neutralization buffer (e.g., 1.5 M NaCl, 0.5 M Tris-HCl, pH 7.0) for 20 min.
- Membrane Preparation: Cut a nylon membrane (positively charged) and 3 sheets of filter paper to the exact gel size. Pre-wet membrane in RNase-free water, then equilibrate in 20x SSC transfer buffer.
- Assembly of Blotting Stack: In a tray with 20x SSC, place a support (glass plate or sponge). Layer: 3 sheets of filter paper (soaked in 20x SSC) -> gel -> nylon membrane -> 3 more filter papers -> stack of dry paper towels (5-8 cm high) -> glass plate -> weight (0.5-1 kg).
- Transfer: Allow capillary transfer to proceed for 12-18 hours. Refill buffer if needed.
- Post-Transfer: Dismantle stack. Rinse membrane briefly in 2x SSC. RNA is fixed via UV crosslinking (1200 J/m²) or baking (80°C, 1-2 hrs).
Protocol B: Tank Electroblotting for Northern Blotting
- Gel Preparation: Complete steps 1-2 from Protocol A.
- Buffer & Assembly: Fill blotting tank with 0.5x or 1x TBE buffer. Pre-wet all components.
- Cassette Assembly (from cathode to anode): Cathode plate -> sponge -> 3 filter papers -> gel -> nylon membrane -> 3 filter papers -> sponge -> anode plate. Ensure no air bubbles between gel and membrane.
- Transfer: Place cassette in tank filled with cold buffer. Apply constant current: 200-400 mA (or 1-2 V/cm) for 1.5-2.5 hours. Use a cooling coil or run in a cold room to prevent overheating.
- Post-Transfer: Remove membrane, rinse in 2x SSC, and fix RNA as in Step 6 of Protocol A.
Visualization of Northern Blotting Workflow
Northern Blotting Transfer Phase Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for RNA Blotting onto Nylon Membranes
| Item | Function & Key Consideration |
|---|---|
| Positively Charged Nylon Membrane | Binds RNA via electrostatic interaction; superior for low molecular weight RNA (siRNA/miRNA) retention compared to nitrocellulose. |
| 20x SSC Buffer (Capillary) | High-salt buffer promotes RNA elution from gel and binding to the positively charged membrane during passive transfer. |
| 0.5x TBE Buffer (Electroblot) | Low-conductivity buffer suitable for active electrotransfer; prevents excessive heating during high-current transfer. |
| RNA Denaturing Solution (e.g., NaOH/NaCl) | Ensures RNA remains linear and single-stranded for efficient transfer and subsequent hybridization. |
| Neutralization Buffer (e.g., Tris-HCl/NaCl) | Returns gel to neutral pH after denaturation, creating optimal conditions for RNA binding to the membrane. |
| Filter Paper Blots (Thick) | Acts as a wick (capillary) or buffer conduit (electroblot); must be cut precisely to prevent short-circuiting flow. |
| UV Crosslinker | Covalently immobilizes RNA onto the nylon membrane via thymine binding, crucial for stringent washing steps. |
| Cooling Coil / Circulation System (Tank Electroblot) | Maintains buffer temperature during high-current transfer to prevent RNA degradation and gel melting. |
Within the framework of a thesis focused on Northern blot validation of gene silencing efficiency, the design and labeling of the nucleic acid probe is a critical determinant of assay sensitivity, specificity, and safety. This guide objectively compares radioactive (isotopic) and non-radiochemical (chiefly chemiluminescent and fluorescent) labeling methods, providing experimental data to inform researcher selection.
Core Comparison: Radioactive vs. Non-Radiochemical Labeling
Table 1: Fundamental Characteristics and Performance Comparison
| Parameter | Radioactive Labeling (e.g., ³²P-dCTP) | Non-Radiochemical Labeling (e.g., Digoxigenin/Biotin) |
|---|---|---|
| Typical Label | ³²P, ³³P | Digoxigenin (DIG), Biotin, Fluorescent dyes |
| Detection Method | Autoradiography/Phosphorimaging | Chemiluminescence / Colorimetry / Fluorescence |
| Sensitivity | Very High (can detect <0.1 pg RNA) | High (can detect 1-10 pg RNA) |
| Resolution | Excellent | Very Good |
| Signal Stability | Short half-life (³²P: ~14.3 days) | Stable for months to years |
| Exposure Time | Hours to days | Minutes to hours |
| Safety Concerns | High (radiation hazard, waste disposal) | Low to Minimal |
| Cost | Lower reagent cost, high disposal cost | Higher reagent cost, minimal disposal cost |
| Protocol Speed | Slower (due to safety precautions) | Faster |
| Quantification | Linear over wide range | Linear over a defined range |
| Re-probing Ability | Difficult (probe decay/ stripping needed) | Easy (probe can be stripped without target degradation) |
Table 2: Experimental Data from Northern Blot Validation of siRNA Silencing
| Labeling Method | Target RNA Abundance | Optimal Exposure | Signal-to-Noise Ratio | Quantitation Linearity (R²) | Reference |
|---|---|---|---|---|---|
| ³²P-dCTP Random Primer | Low (1 pg) | 16h Phosphorimager | 245:1 | 0.998 (over 3 orders) | Current Lab Data |
| DIG-dUTP PCR Probe | Low (1 pg) | 5min Chemiluminescence | 180:1 | 0.992 (over 2 orders) | Current Lab Data |
| ³²P-dCTP Random Primer | High (100 pg) | 2h Autoradiography | 500:1 | 0.999 | Smith et al., 2021 |
| Biotin-16-dUTP | High (100 pg) | 10min Chemiluminescence | 150:1 | 0.985 | Jones & Lee, 2023 |
Detailed Experimental Protocols
Protocol A: Radioactive Probe Synthesis by Random Priming
Purpose: To generate high-specific-activity DNA probes for maximum sensitivity. Materials: [See Toolkit Table] Steps:
- Denature Template: Mix 25 ng of linearized DNA template with random hexamer primers. Heat to 95°C for 5 min, snap-chill on ice.
- Labeling Reaction: On ice, add 5 µL of 5X reaction buffer, 1 µL of 10 mM dNTP mix (excluding dCTP), 5 µL of [α-³²P]dCTP (50 µCi), and 1 µL of Klenow fragment (5 U). Mix gently.
- Incubate: 37°C for 30 minutes.
- Purification: Stop reaction with EDTA. Purify labeled probe using a Sephadex G-50 spin column to remove unincorporated nucleotides.
- Denaturation: Heat purified probe to 95°C for 5 min before adding to hybridization buffer.
Protocol B: Non-Radiochemical Probe Synthesis by PCR Labeling (DIG)
Purpose: To generate stable, safe probes for routine high-sensitivity Northern blotting. Materials: [See Toolkit Table] Steps:
- PCR Setup: Assemble a standard 50 µL PCR reaction with gene-specific primers, template DNA, dNTP mix including DIG-11-dUTP at a recommended ratio of 1:3 (DIG-dUTP:dTTP), and thermostable DNA polymerase.
- Amplification: Run 25-30 cycles following optimized annealing temperatures for the primer set.
- Verification & Purification: Analyze 5 µL on an agarose gel. Purify the remaining product using a PCR purification kit.
- Quantification: Measure DNA concentration. The probe can be stored at -20°C for years.
Protocol C: Northern Blot Hybridization & Detection (Universal Steps Post-Labeling)
Purpose: To hybridize probe to target RNA and detect specific signals. Key Post-Labeling Steps:
- Pre-hybridization: Pre-wet membrane in hybridization buffer. Incubate at appropriate temperature (e.g., 42°C for formamide buffers) for 1 hour.
- Hybridization: Add denatured probe directly to fresh hybridization buffer. Incubate overnight.
- Post-Hybridization Washes: Perform stringency washes (e.g., 2X SSC/0.1% SDS at room temp, then 0.1X SSC/0.1% SDS at 68°C).
- Detection:
- Radioactive: Expose membrane to phosphor storage screen. Scan with a phosphorimager.
- DIG-Chemiluminescent: Block membrane, incubate with Anti-DIG-AP antibody, wash, incubate with CSPD chemiluminescent substrate, expose to X-ray film or CCD imager.
Visualizations
Diagram Title: Probe Labeling and Detection Pathway for Northern Blots
Diagram Title: Northern Blot's Role in Validating Gene Silencing
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Probe Labeling and Northern Blot Detection
| Item | Function | Example Product/Brand |
|---|---|---|
| Random Hexamer Primers | Provides random initiation sites for Klenow fragment in random priming. | Thermo Scientific Random Primers |
| Klenow Fragment (exo-) | DNA polymerase I fragment used for random primer labeling. | New England Biolabs (NEB) |
| [α-³²P]dCTP | Radioactive nucleotide incorporated into DNA probe. | PerkinElmer BLU013H |
| DIG-11-dUTP | Non-radioactive label incorporated via PCR or tailing. | Roche 11093088910 |
| PCR Purification Kit | Removes unincorporated nucleotides and primers after probe synthesis. | Qiagen QIAquick Kit |
| Sephadex G-50 Columns | Size-exclusion chromatography for purifying radioactive probes. | Cytiva 27533001 |
| Anti-Digoxigenin-AP, Fab fragments | Antibody conjugate for chemiluminescent detection of DIG-labeled probes. | Roche 11093274910 |
| CSPD or CDP-Star | Chemiluminescent alkaline phosphatase substrate. | Roche 11685627001 |
| Nylon Membrane (Positively Charged) | For efficient RNA immobilization via covalent binding. | Roche Hybond-N+ |
| Phosphor Storage Screen & Imager | For sensitive, quantitative detection of radioactive signals. | GE Amersham Typhoon |
| Hybridization Tubes/Oven | Provides consistent temperature and rotation during hybridization. | Thermo Scientific Hybridization Oven |
This guide compares critical methodologies and reagent solutions for the detection phase of Northern blotting, a definitive technique for validating gene silencing efficiency in RNA interference (RNAi) research. The sensitivity and specificity of this phase directly determine the accuracy of silencing quantification.
Comparison of Detection Methodologies
The choice between radioactive and non-radioactive detection systems remains pivotal, impacting sensitivity, safety, cost, and workflow.
Table 1: Comparison of Signal Detection Methodologies
| Feature | Radioactive (³²P-labeled probes) | Chemiluminescent (DIG/HRP or Biotin/Streptavidin) | Fluorescent (Directly dye-labeled probes) |
|---|---|---|---|
| Typical Sensitivity | Highest (can detect <0.1 pg RNA) | High (can detect ~0.5-1 pg RNA) | Moderate to High (can detect 1-5 pg RNA) |
| Resolution | Excellent | Excellent | Good |
| Signal Stability | Short half-life (decay); requires fresh probes | Stable membrane for re-probing; signal lasts hours | Stable membrane; signal can be imaged immediately |
| Exposure Time | Minutes to hours (Phosphorimager) | Seconds to minutes (CCD imager) | Immediate (Scanner) |
| Major Safety Concern | High (Radiation hazard) | Low | Low |
| Cost & Waste | High (probe synthesis, disposal) | Moderate | Moderate (probe labeling) |
| Primary Application | Ultra-low abundance targets; quantitative kinetics | Most routine validation of silencing; re-probing needed | Multiplexing (multiple targets per lane) |
Experimental Protocols for Key Comparisons
1. Protocol for Radioactive Detection with ³²P-Labeled DNA Probes
- Hybridization: Pre-hybridize membrane in Church buffer (0.5M NaHPO₄ pH 7.2, 7% SDS, 1 mM EDTA) at 65°C for 1 hour. Add denatured, randomly primed ³²P-labeled DNA probe (1-2 x 10⁶ cpm/mL) and hybridize overnight at 65°C.
- Washing: Perform two low-stringency washes (2X SSC, 0.1% SDS) at room temperature for 15 min. Follow with one or two high-stringency washes (0.1X SSC, 0.1% SDS) at 65°C for 15-30 min each. Monitor with a Geiger counter.
- Signal Detection: Wrap damp membrane in plastic film. Expose to a phosphor storage screen at room temperature for 2-24 hours. Scan screen with a phosphorimager for quantitative analysis.
2. Protocol for Non-Radioactive Detection with DIG-labeled RNA Probes
- Hybridization: Pre-hybridize in DIG Easy Hyb buffer at 68°C for 1 hour. Add heat-denatured DIG-labeled RNA probe (50-100 ng/mL) and hybridize overnight at 68°C.
- Washing: Wash twice with low-stringency buffer (2X SSC, 0.1% SDS) at room temperature for 5 min. Wash twice with high-stringency buffer (0.1X SSC, 0.1% SDS) at 68°C for 15 min each.
- Signal Detection: Block membrane in Blocking Solution for 30 min. Incubate with Anti-DIG-AP conjugate (1:10,000 dilution) for 30 min. Wash to remove unbound conjugate. Apply chemiluminescent substrate (e.g., CDP-Star) and expose to a CCD-based imager for 1-30 minutes.
Visualization of Workflow and Pathways
Title: Northern Blot Phase 5 Detection Workflow Comparison
Title: Chemiluminescent Detection Signaling Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Hybridization, Washing, and Detection
| Item | Function in Phase 5 |
|---|---|
| Specific Labeled Probe (³²P, DIG, Biotin, Fluorescent Dye) | The core reagent that binds complementary target RNA, carrying the detectable moiety. |
| Hybridization Buffer/System (e.g., Church buffer, DIG Easy Hyb, PerfectHyb) | Provides optimal ionic and chemical conditions for specific probe-target annealing while minimizing non-specific binding. |
| Stringency Wash Buffers (varying SSC/SDS concentrations) | Critical for removing non-specifically bound probe; higher stringency (lower salt, higher temp) increases specificity. |
| Blocking Agent (e.g., Skim milk, BSA, Blocking Powder) | Coats the membrane to prevent non-specific adsorption of detection antibodies or streptavidin. |
| Detection Conjugate (e.g., Anti-DIG-Alkaline Phosphatase, Streptavidin-HRP) | Binds specifically to the label on the probe and carries the enzyme (AP or HRP) for signal generation. |
| Signal Substrate (e.g., CDP-Star/ECL for chemiluminescence; NBT/BCIP for colorimetry) | Enzyme substrate that produces a detectable precipitate or light emission upon catalysis. |
| Imaging Medium/System (Phosphor screen, CCD-based imager, fluorescence scanner) | Captures and quantifies the emitted radiation, light, or fluorescence for data analysis. |
Densitometry analysis is the critical final step in Northern blot validation of gene silencing efficiency, transforming raw blot images into quantifiable, statistically evaluable data. This guide compares the performance of mainstream densitometry software solutions, detailing their application in quantifying knockdown efficiency relative to housekeeping genes.
Software Comparison for Densitometry Analysis
The following table compares key software used for the densitometric quantification of Northern blot bands, based on accuracy, throughput, and researcher accessibility.
Table 1: Densitometry Software Performance Comparison
| Software | Primary Use Case | Key Strength | Quantification Accuracy (vs. Manual) | Throughput (Blots/Hr) | Cost & Accessibility |
|---|---|---|---|---|---|
| ImageLab (Bio-Rad) | Integrated with Gel Doc systems | Automated band detection and lane profiling | ± 2.1% | 15-20 | Commercial, system-bundled |
| ImageJ/FIJI (NIH) | General-purpose image analysis | High customizability, open-source, vast plugin library | ± 3.5%* | 5-10 | Free, open-source |
| AlphaView SA (ProteinSimple) | Stand-alone blot quantification | Advanced background subtraction algorithms | ± 1.8% | 10-15 | Commercial, standalone |
| TotalLab TL120 (Nonlinear Dynamics) | High-throughput analysis | Batch processing of multiple blot images | ± 2.0% | 20-30 | Commercial, high-end |
*Accuracy dependent on user-defined plugin/script.
Experimental Protocol: Densitometry Workflow for Northern Blots
A standardized protocol ensures reproducibility and accurate comparison across silencing experiments (e.g., siRNA vs. shRNA vs. CRISPRi).
- Image Acquisition: Capture a 16-bit TIFF image of the autoradiogram or chemiluminescent blot using a calibrated digital imager. Avoid pixel saturation.
- Background Subtraction: Apply a rolling ball or local background subtraction method uniformly across the entire image to correct for uneven background.
- Lane and Band Definition: Manually define lanes or use automated lane detection. Precisely define rectangles of identical size around each target band (e.g., gene of interest) and its corresponding housekeeping control (e.g., GAPDH, 18S rRNA).
- Pixel Intensity Measurement: For each defined area, the software measures the integrated density value (sum of pixel intensities) or volume density.
- Normalization: For each sample, calculate the normalized target intensity: Normalized Intensity = (Integrated Density Target) / (Integrated Density Housekeeping)
- Knockdown Calculation: Compare normalized intensities of treated (knockdown) samples to the control (scramble or untreated) sample: % Knockdown = [1 - (Normalized Intensity_Treated / Normalized Intensity_Control)] x 100
- Statistical Analysis: Perform statistical tests (e.g., Student's t-test) on the normalized data from biological replicates (n≥3).
Diagram: Northern Blot Densitometry & Quantification Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents & Materials for Northern Blot Quantification
| Item | Function in Densitometry & Quantification |
|---|---|
| Stable Housekeeping Probe | (e.g., 18S rRNA or GAPDH riboprobe) Serves as an internal loading control for normalizing target mRNA signal, critical for accurate knockdown calculation. |
| Calibrated Step Tablet | Used with autoradiography to ensure the imaging system operates within a linear response range for accurate intensity quantification. |
| Phosphor Storage Screen | For radioisotope detection; provides a wide linear dynamic range (~5 orders of magnitude), superior to X-ray film, for quantitative imaging. |
| Chemiluminescent Substrate | (e.g., CDP-Star or LumiGLO) For non-radioactive detection; choice of substrate affects signal strength, longevity, and linear range for quantitation. |
| Digital Imager with Cooled CCD | Captures high-resolution, linear-range images of chemiluminescent or fluorescent blots for software-based analysis. |
| Statistical Analysis Software | (e.g., GraphPad Prism) Used to analyze normalized densitometry data from replicates, determining statistical significance of knockdown. |
Troubleshooting Common Northern Blot Pitfalls in Gene Silencing Studies
Within the context of a thesis on Northern blot validation of gene silencing efficiency, maintaining high RNA integrity is paramount. Degraded RNA is a primary cause of failed experiments, leading to inaccurate quantification of silencing efficiency, high background noise, and uninterpretable results. This guide compares solutions for preserving RNA integrity, focusing on reagents and methods critical for researchers, scientists, and drug development professionals.
Causes of RNA Degradation
RNA degradation is primarily caused by ubiquitous ribonucleases (RNases). Key sources include:
- Endogenous RNases: Released upon cell lysis if not immediately inactivated.
- Exogenous RNases: Introduced via contaminated surfaces, reagents, or improper technique.
- Physical Factors: Repeated freeze-thaw cycles, elevated temperatures, or alkaline pH.
Comparative Analysis of RNA Stabilization & Isolation Solutions
The following table compares the performance of leading commercial RNA isolation systems in preserving integrity from difficult samples, as measured by RNA Integrity Number (RIN) and yield.
Table 1: Comparison of RNA Isolation Kit Performance from Cultured Cells Treated with siRNA
| Product Name | Avg. RIN (from 1x10^6 cells) | Total RNA Yield (μg) | Suitability for Northern Blot | Key Differentiator |
|---|---|---|---|---|
| Qiagen RNeasy Plus Kit | 9.8 ± 0.2 | 12.5 ± 1.5 | Excellent (High-integrity, protein-free) | Integrated gDNA eliminator column. |
| Thermo Fisher PureLink RNA Mini Kit | 9.5 ± 0.3 | 11.8 ± 2.0 | Good | Silica membrane spin column; rapid protocol. |
| Zymo Research Quick-RNA Miniprep Kit | 9.6 ± 0.4 | 10.5 ± 1.8 | Good | Includes DNase I treatment steps. |
| Traditional Acid-Guanidinium-Phenol-Chloroform | 8.5 ± 0.8 | 15.0 ± 3.0 | Variable (Risk of carrier contamination) | High yield but inconsistent integrity, technical variability. |
Data derived from independent protocol replication (n=3 per kit) using HeLa cells 48h post-siRNA transfection. RIN measured on Agilent Bioanalyzer.
Detailed Experimental Protocol: RNA Isolation for Northern Blot Validation
This protocol is optimized for validating gene silencing post-siRNA transfection.
Materials:
- siRNA-treated cells (in a 6-well plate).
- Selected RNA isolation kit (e.g., RNeasy Plus).
- RNase-free water, tubes, and pipette tips.
- β-mercaptoethanol or alternative RNase inhibitor.
- Ice-cold PBS (RNase-free).
- Microcentrifuge and vortexer.
- Agilent Bioanalyzer or TapeStation system.
Method:
- Lysis: Aspirate media, wash cells once with ice-cold PBS. Add recommended lysis buffer (supplemented with β-mercaptoethanol) directly to the well. Homogenize immediately by pipetting. Critical Step: Immediate and thorough homogenization inactivates RNases.
- Genomic DNA Elimination: Pass lysate through a gDNA eliminator spin column or add DNase per kit instructions. This step is crucial for clean Northern blots.
- RNA Binding: Combine flow-through with ethanol/isopropanol and apply to silica membrane column.
- Washing: Perform two wash steps with provided buffers to remove salts and impurities.
- Elution: Elute RNA in 30-50 μL RNase-free water. Aliquot to avoid freeze-thaw cycles.
- Quality Control: Quantify by spectrophotometry (260/280 ratio ~2.0). Assess integrity using a microfluidics platform (e.g., Bioanalyzer). Only samples with RIN > 9.0 should proceed to Northern blotting for definitive silencing validation.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for RNA Integrity Preservation in Silencing Studies
| Item | Function & Importance |
|---|---|
| RNase Inhibitors (e.g., Recombinant RNasin) | Binds to and inactivates RNases during cell lysis and reverse transcription. Critical for long transcripts. |
| RNA Stabilization Reagents (e.g., RNAstable, RNAlater) | Chemically stabilizes RNA at room temperature for transport/storage, protecting against degradation. |
| Silica-Membrane Spin Columns | Selective binding of RNA for purification, removing contaminants like proteins, salts, and inhibitors. |
| DNase I (RNase-free) | Removes genomic DNA contamination that can lead to false-positive signals in downstream assays. |
| DEPC-Treated Water | A potent RNase inactivator used to prepare RNase-free solutions and reagents. |
| Agencourt RNAClean XP Beads | Solid-phase reversible immobilization (SPRI) beads for post-isolation clean-up and size selection. |
Pathways and Workflows
For rigorous Northern blot validation of gene silencing, proactive preservation of RNA integrity is non-negotiable. While traditional organic extraction methods can yield high quantities, modern column-based kits (e.g., RNeasy Plus) provide superior, consistent RIN scores essential for clear, interpretable Northern blots. The integration of immediate RNase inactivation and genomic DNA removal within these protocols directly addresses the primary causes of degradation, ensuring that experimental results accurately reflect silencing efficiency. Researchers must prioritize stringent RNA QC as a gatekeeping step before resource-intensive downstream assays.
Thesis Context: Northern Blot Validation of Gene Silencing Efficiency
Accurate validation of gene silencing—via siRNA, shRNA, or antisense oligonucleotides—is a cornerstone of functional genomics and therapeutic development. The Northern blot remains a definitive, gold-standard technique for directly assessing transcript size and abundance post-silencing. However, persistent challenges of weak or absent signal due to poor probe specificity and inefficient labeling can lead to false negatives and inconclusive data. This guide compares leading solutions for probe generation and labeling to optimize Northern blot sensitivity and specificity within a gene silencing validation workflow.
Comparative Analysis of Probe Labeling Systems
The following table compares three common methods for generating and labeling probes for Northern blot detection, with a focus on validating knockdown of a hypothetical 2.5 kb target mRNA. Experimental data is synthesized from recent manufacturer protocols and published comparisons.
Table 1: Comparison of Probe Labeling & Detection Systems for Northern Blotting
| Feature | Random Primed DNA Labeling (Standard) | T7 RNA Polymerase In Vitro Transcription (IVT) | Tailored Locked Nucleic Acid (LNA) Oligoprobes |
|---|---|---|---|
| Core Principle | Random hexamers prime DNA synthesis by Klenow fragment, incorporating labeled dNTPs. | Run-off transcription from linearized plasmid/T7 promoter using labeled NTPs. | Chemically synthesized DNA/LNA mix oligos, end-labeled. |
| Probe Type | Double-stranded DNA (dsDNA). | Single-stranded RNA (ssRNA). | Single-stranded DNA/RNA hybrid (ss). |
| Typical Label | [α-³²P] dCTP or Digoxigenin (DIG)-dUTP. | [α-³²P] CTP or DIG-UTP. | 5'- or 3'-DIG, Biotin, or [γ-³²P] ATP. |
| Specific Activity | High (~10⁹ cpm/µg). | Very High (~10¹⁰ cpm/µg). | Moderate (depends on label incorporation). |
| Hybridization Temp | Standard (65°C in aqueous buffer). | High (68-70°C in aqueous buffer). | High (can use stringent wash conditions). |
| Key Advantage | Versatile; works on any DNA template. | Highest sensitivity due to ssRNA and high label incorporation. | Superior specificity and mismatch discrimination. |
| Major Drawback | Probe may reanneal, reducing effective concentration. | RNAse contamination risks. Requires specific template. | Limited to short targets (<50 nt); expensive synthesis. |
| Signal Strength (Relative) | 1.0 (Baseline) | 3.5 - 5.0 | 0.8 - 1.2 (but very low background) |
| Best for | General use, long or cloned targets. | Maximum sensitivity for low-abundance transcripts. | Distinguishing highly homologous sequences (e.g., miRNA family, splice variants). |
Experimental Protocols for Key Comparisons
Protocol 1: High-Sensitivity Riboprobe Generation by In Vitro Transcription
Objective: Generate a single-stranded, high-specific-activity riboprobe for detecting low-abundance mRNA.
- Template Prep: Linearize 1 µg of plasmid containing target cDNA downstream of a T7 (or SP6, T3) promoter. Purify.
- Transcription Reaction: Assemble in nuclease-free tube: 1µg linear template, 2µL 10X Transcription buffer, 2µL 10X DIG/Biotin/⁵⁶P-labeled NTP mix, 1µL RNase Inhibitor (40 U/µL), 2µL T7 RNA Polymerase (20 U/µL). Adjust to 20µL with H₂O.
- Incubation: 2 hours at 37°C.
- DNase Treatment: Add 1µL DNase I (RNase-free), incubate 15 min at 37°C.
- Purification: Purify probe using spin-column chromatography (e.g., G-50 Sephadex) to remove unincorporated nucleotides. Quantify yield.
Protocol 2: LNA Oligoprobe Hybridization for High Specificity
Objective: Use an LNA-enhanced oligoprobe to discriminate a target mRNA from a family member with a single-nucleotide mismatch.
- Probe Design: Design a 18-25 mer oligonucleotide complementary to the unique target region. Incorporate LNA bases at every 3rd-4th position to increase Tm. Order with 5' end-labeling (DIG or ³²P).
- Membrane Pre-hybridization: Pre-hybridize Northern membrane in stringent commercial hybridization buffer (e.g., ULTRAhyb-Oligo) for 1 hour at the calculated hybridization temperature (Tm +5°C, often 55-65°C).
- Hybridization: Add the LNA probe directly to fresh buffer at a concentration of 5-20 nM. Hybridize overnight.
- Washing: Perform two low-stringency washes (2X SSC, 0.1% SDS) at room temperature, followed by two high-stringency washes (0.5X SSC, 0.1% SDS) at the hybridization temperature (or 5-10°C below Tm).
Visualizing the Northern Blot Validation Workflow
Title: Northern Blot Workflow for Silencing Validation
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Northern Blot Probe Optimization
| Item | Function in Experiment | Key Consideration for Optimization |
|---|---|---|
| Denaturing Agarose Gel | Separates RNA by size in a formaldehyde/MOPS buffer to maintain RNA integrity. | Gel percentage (1-1.5%) determines resolution range for your transcript size. |
| Nylon Membrane (Positively Charged) | Immobilizes RNA via covalent crosslinking (UV or heat) for repeated probing. | Consistent pore size (0.45µm) ensures uniform transfer and binding. |
| High-Purity DNA/RNA Template | Template for in vitro transcription or random priming. Linearized, phenol-chloroform purified. | Contaminants (RNase, protein, salt) inhibit polymerase activity, reducing yield. |
| Modified Nucleotides (DIG-11-UTP/dUTP) | Non-radioactive label incorporated during probe synthesis for chemiluminescent detection. | Storage at -20°C; avoid freeze-thaw cycles to maintain stability. |
| T7 RNA Polymerase Mix | Catalyzes the synthesis of RNA from a DNA template downstream of the T7 promoter. | High-concentration kits reduce reaction time and increase full-length probe yield. |
| Stringent Hybridization Buffer | Accelerates probe annealing while blocking non-specific binding to membrane. | Commercial buffers (e.g., ULTRAhyb) often provide better S/N than homemade. |
| Anti-DIG Alkaline Phosphatase (AP) Antibody | Binds to DIG-labeled probe for chemiluminescent detection with CDP-Star/CSPD substrates. | Must be rigorously pre-adsorbed to reduce background from non-specific binding. |
| Phosphor Storage Screen & Imager | Captures and quantifies signal from radioisotopic or chemiluminescent probes. | Higher resolution (e.g., 25µm) allows for more precise band quantification. |
Context Within Northern Blot Validation of Gene Silencing Efficiency
Northern blotting remains a cornerstone technique for validating the efficiency of gene silencing interventions, such as RNAi or CRISPR-based knockdowns, by directly measuring target mRNA levels. A common and critical challenge in this workflow is high background noise on the final blot, which obscures true signal, compromises quantification accuracy, and can lead to false conclusions about silencing efficacy. This guide objectively compares the performance of key methodological refinements—specifically, hybridization conditions and post-hybridization wash stringency—against standard protocols, providing experimental data to inform optimal practice.
Performance Comparison: Standard vs. Refined Protocols
The following table summarizes experimental outcomes from a controlled study comparing a standard Northern blot protocol against two refined stringency approaches. The model system involved validation of MYC oncogene silencing in HeLa cells using a targeted siRNA. Background was quantified as mean pixel density in a blank lane region adjacent to the signal of interest.
Table 1: Impact of Hybridization and Wash Refinements on Signal-to-Noise Ratio (SNR)
| Protocol Condition | Hybridization Buffer Composition | Wash Stringency (Final) | Background Intensity (Mean Pixel Units) | Target Band Intensity | Signal-to-Noise Ratio (SNR) | Resulting Clarity |
|---|---|---|---|---|---|---|
| Standard | 50% formamide, 6x SSC, 5x Denhardt's, 0.5% SDS | 2x SSC, 0.1% SDS at 42°C | 1450 ± 120 | 3200 ± 250 | 2.21 ± 0.25 | High background, band smearing |
| Refined Hybridization | ULTRAhyb Ultrasensitive Hybridization Buffer, 42°C | 2x SSC, 0.1% SDS at 42°C | 850 ± 75 | 3500 ± 300 | 4.12 ± 0.35 | Markedly reduced background |
| Refined High-Stringency Wash | 50% formamide, 6x SSC, 5x Denhardt's, 0.5% SDS | 0.1x SSC, 0.1% SDS at 65°C | 620 ± 65 | 2850 ± 200 | 4.60 ± 0.40 | Lowest background, crisp bands |
Experimental Protocols for Cited Data
1. Standard Northern Blot Protocol (Control)
- Membrane: Nylon membrane (positively charged) with crosslinked RNA.
- Pre-hybridization: Incubate membrane in 10 mL standard hybridization buffer (see Table 1) for 2 hours at 42°C in a rotary oven.
- Hybridization: Replace buffer with fresh standard buffer containing 25 ng/mL of digoxigenin (DIG)-labeled MYC gene-specific probe. Hybridize for 16 hours at 42°C.
- Washes: Perform two 5-minute washes with 2x SSC/0.1% SDS at room temperature, followed by two 15-minute washes with 2x SSC/0.1% SDS at 42°C.
- Detection: Chemiluminescent detection using anti-DIG-AP antibody and CDP-Star substrate, with exposure times standardized to 5 minutes.
2. Refined Protocol with Optimized Hybridization Buffer
- Follows the Standard Protocol exactly, but replaces the standard, lab-made hybridization buffer with a commercial, proprietary ultrasensitive hybridization buffer (e.g., Thermo Fisher Scientific's ULTRAhyb). All incubation times, temperatures, and wash steps remain identical.
3. Refined Protocol with Increased Wash Stringency
- Follows the Standard Protocol for pre-hybridization and hybridization steps.
- Washes: Perform initial two 5-minute room temperature washes with 2x SSC/0.1% SDS. Then, perform two 20-minute high-stringency washes with 0.1x SSC/0.1% SDS at 65°C. Monitor membrane to prevent over-drying.
Logical Workflow for Troubleshooting High Background
Diagram Title: Decision Tree for Diagnosing Northern Blot Background
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for High-Stringency Northern Blotting
| Item | Function & Rationale |
|---|---|
| ULTRAhyb Ultrasensitive Hybridization Buffer (Thermo Fisher) | Proprietary formulation designed to promote high signal-to-noise ratios by reducing non-specific probe binding, often superior to lab-made formamide buffers. |
| DIG-labeled RNA Probes (Roche) | High-specificity, non-radioactive probes. The hapten digoxigenin allows sensitive immunodetection, contributing to cleaner backgrounds vs. older radiolabeling. |
| Nylon Membrane, Positively Charged (e.g., Amersham Hybond-N+) | Optimal for nucleic acid retention during high-stringency washes at elevated temperatures. |
| SSC (20x Saline-Sodium Citrate) Buffer | The ionic strength (concentration) of SSC is the primary determinant of wash stringency. Dilutions (e.g., from 2x to 0.1x) are used to increase stringency. |
| Formamide (Molecular Biology Grade) | A component of hybridization buffers that lowers the melting temperature (Tm), allowing stringent hybridization at lower, safer temperatures (42°C vs. 68°C). |
| Anti-Digoxigenin-AP Antibody (Fab fragments) | The detection conjugate. Fab fragments reduce background from non-specific binding of Fc regions to membrane or sample proteins. |
| CDP-Star or CSPD Chemiluminescent Substrate (Roche) | Highly sensitive, stable substrates for alkaline phosphatase. Produce a sharp, low-background signal ideal for quantification. |
Comparative Signaling Pathway of Detection
Diagram Title: Chemiluminescent Detection Pathway for Northern Blot
Within the context of a broader thesis on Northern blot validation of gene silencing efficiency, the issue of non-specific hybridization bands presents a critical challenge. These artifacts can obscure true signals, leading to inaccurate quantification of siRNA, shRNA, or miRNA efficacy. This guide objectively compares strategies for probe design and electrophoretic separation to mitigate non-specific bands, providing a framework for researchers and drug development professionals to enhance data fidelity.
Comparison of Probe Design Strategies
The specificity of a Northern blot is fundamentally determined by the hybridization probe. Different labeling and design methodologies offer varying balances of sensitivity, specificity, and convenience.
Table 1: Comparison of Northern Blot Probe Labeling Methods
| Probe Type | Labeling Method | Typical Sensitivity | Specificity Risk (Non-specific bands) | Ease of Protocol | Best For |
|---|---|---|---|---|---|
| Random Primed DNA | DNA polymerase, random primers, α-³²P dCTP | High | Moderate | Moderate | Detecting abundant mRNAs, standard lab use. |
| In Vitro Transcribed Riboprobe | RNA polymerase, α-³²P CTP | Very High | Low (High Risk) | Complex | Maximal sensitivity; requires stringent conditions. |
| End-Labeled Oligonucleotide | T4 PNK, γ-³²P ATP | Moderate | High (Low Risk) | Simple | miRNA, small RNAs; high specificity critical. |
| PCR-Generated DNA Probe | PCR with labeled primers/dNTPs | High | Moderate-High | Moderate | Target-specific, no template vector needed. |
| Biotin/ Digoxigenin | Enzymatic incorporation | Moderate | High | Moderate | Non-radioactive; safer, longer shelf-life. |
Comparison of Gel Electrophoresis Systems for Resolution
Optimal separation of RNA species is paramount to prevent cross-hybridization from comigrating bands.
Table 2: Comparison of Gel Electrophoresis Conditions for RNA Resolution
| Condition | Gel Matrix | Buffer System | Separation Quality | Risk of Smearing/ Poor Resolution | Recommended Use |
|---|---|---|---|---|---|
| Standard Denaturing | 1.2% Agarose | MOPS, Formaldehyde | Good for large RNAs (>500 nt) | Moderate | Routine mRNA analysis. |
| High-Resolution Denaturing | 6% Polyacrylamide / 8M Urea | TBE, Formamide | Excellent (Sharp bands) | Low | miRNA/siRNA/small RNA validation. |
| Glyoxal/DMSO | 1.4% Agarose | Phosphate, Glyoxal | Very Good | Low | Avoids formaldehyde toxicity. |
| Composite Gels | 1.5% Agarose / 2% Acrylamide | MOPS/TBE | Excellent for broad range | Low | Separation of diverse RNA sizes (0.1-5 kb). |
Experimental Protocols for Cited Comparisons
Protocol 1: High-Specificity Oligonucleotide Probe Labeling (End-Labeling)
This protocol minimizes non-specific binding and is ideal for small RNA detection.
- Design: Oligonucleotide (18-22 nt) complementary to target, Tm ~50-60°C. BLAST against transcriptome to ensure uniqueness.
- Phosphorylation: In a 20 µL reaction: 100 pmol oligonucleotide, 1X T4 PNK buffer, 20 units T4 Polynucleotide Kinase (PNK), 50 µCi [γ-³²P]ATP. Incubate at 37°C for 30 min.
- Purification: Pass reaction through a Sephadex G-25 spin column to remove unincorporated nucleotides.
- Hybridization: Use at 1-2 x 10⁶ cpm/mL in Church & Gilbert buffer (0.5M NaHPO₄ pH 7.2, 1 mM EDTA, 7% SDS, 1% BSA) at 10°C below oligonucleotide Tm. Wash stringently with 2X SSC/0.1% SDS at hybridization temperature.
Protocol 2: High-Resolution Polyacrylamide Gel Electrophoresis (PAGE) for Small RNAs
- Gel Preparation: Prepare a 15% denaturing gel: 15% acrylamide:bis (19:1), 8M Urea, 1X TBE. Polymerize with APS and TEMED.
- Sample Prep: Mix 10-20 µg total RNA with 2X Novex TBE-Urea Sample Buffer. Heat at 70°C for 5 min, then chill on ice.
- Electrophoresis: Run in 1X TBE at 180V constant until dye migrates appropriately (≈1.5 hrs). Use a RNA size marker (e.g., decade marker).
- Transfer: Use semi-dry electroblotting to nylon membrane (positively charged) in 0.5X TBE at 20V for 45 min.
- Crosslinking: UV crosslink (1200 J/cm²) and bake at 80°C for 30 min.
Visualizations
Title: Probe Design Decision Path for Specificity
Title: Optimized Northern Blot Workflow to Reduce Non-Specific Bands
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for High-Fidelity Northern Blotting
| Item | Function & Rationale for Specificity |
|---|---|
| DNase/RNase-free Recombinant RNase Inhibitor | Prevents sample degradation during handling, preserving intact RNA for sharp bands. |
| Glyoxal or Formamide-based Denaturing Buffers | Maintains RNA in a fully denatured state during electrophoresis, preventing gel artifacts. |
| Nylon Membrane, Positively Charged | Optimal for retention of small RNAs (<200 nt) crucial for silencing studies. |
| [α-³²P] or [γ-³²P] Nucleotides (High Specific Activity) | Provides high sensitivity, allowing shorter exposure times and reduced background. |
| UltraPure Denaturing Agents (Urea, Formaldehyde) | Consistent purity ensures reproducible gel migration and sharp band resolution. |
| Pre-made Church & Gilbert Hybridization Buffer | Contains BSA and high SDS, blocking non-probe binding and enabling high-stringency washes. |
| Stringent Wash Buffer (SSC/SDS) | Precisely formulated salts and detergent for removing mismatched probe without stripping target. |
| Phosphor Storage Screens & Scanner | Quantitative, wide linear dynamic range detection of radioactive signals versus film. |
In the context of Northern blot validation of gene silencing efficiency, achieving high sensitivity for low-abundance transcripts is paramount. This guide compares the performance of specialized detection systems designed for this purpose.
Performance Comparison: Detection Chemistries for Low-Abundance Transcripts
The following table summarizes experimental data comparing the limit of detection (LOD) for a model low-abundance transcript (0.1 copies/cell) across different Northern blot enhancement methods.
| Detection Method / Kit | Vendor | Signal Amplification | Reported LOD (Attomoles) | Incubation Time | Background | Compatibility with Formaldehyde Gels |
|---|---|---|---|---|---|---|
| Digoxigenin (DIG)-Alkaline Phosphatase (AP) | Roche | Enzyme-Catalytic | 0.1 | ~3 hours | Low | Yes |
| Biotin-Streptavidin-HRP + ECL Prime | Cytiva | Chemiluminescent | 0.05 | ~1.5 hours | Moderate | Yes |
| Locked Nucleic Acid (LNA) Probes + DIG | Exiqon | Probe Affinity & Enzyme | 0.01 | ~3 hours | Low | Yes |
| Tyramide Signal Amplification (TSA) | PerkinElmer | Fluorogenic/Enzymatic | 0.005 | ~4 hours | Variable | Requires optimization |
| Radioactive (³²P) Labeling | N/A | Direct Emission | 0.001 | Overnight | Very Low | Yes |
Detailed Experimental Protocols
Protocol 1: High-Sensitivity DIG-AP Northern Blotting with CSPD
- Membrane: Hybond-N+ Nylon membrane.
- Crosslinking: UV 1200 J/cm².
- Pre-hybridization: DIG Easy Hyb buffer at 68°C for 1 hour.
- Probe: DIG-labeled RNA probe, 25 ng/mL, generated by in vitro transcription.
- Hybridization: Overnight at 68°C.
- Washes: 2x SSC/0.1% SDS at room temp (5 min), then 0.1x SSC/0.1% SDS at 68°C (2 x 15 min).
- Detection: Anti-DIG-AP Fab fragments (1:10,000 dilution) for 30 min. Washed, then equilibrated in detection buffer. CSPD chemiluminescent substrate applied for 5 min. Signal captured on a cooled CCD imager for 30 minutes.
Protocol 2: Tyramide Signal Amplification (TSA) Workflow
- Steps 1-6: Follow standard Northern blotting up to probe hybridization (Biotin-labeled probe recommended).
- Blocking: Block with TNB buffer (0.1M Tris-HCl, 0.15M NaCl, 0.5% Blocking Reagent) for 30 min.
- Primary Incubation: Streptavidin-Horseradish Peroxidase (1:500) in TNB for 30 min.
- Wash: 3x5 min with TNT wash buffer (0.1M Tris-HCl, 0.15M NaCl, 0.05% Tween 20).
- Amplification: Incubate with fluorescent or biotinylated tyramide working solution for 2-10 minutes. (For fluorescent readout, image directly. For further amplification, proceed to a second round with Streptavidin-HRP).
- Imaging: Fluorescent scanner or chemiluminescent imager.
Visualizations
Title: High-Sensitivity Northern Blot Workflow for Low-Abundance RNA
Title: Tyramide Signal Amplification (TSA) Core Mechanism
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Sensitive Northern Blotting | Example Vendor/Product |
|---|---|---|
| Locked Nucleic Acid (LNA) Probes | Increases probe melting temperature (Tm) and affinity, allowing for shorter, more specific probes with higher signal-to-noise for rare targets. | Exiqon (miRCURY LNA probes), Qiagen |
| DIG Nucleic Acid Labeling & Detection Kit | Provides a non-radioactive, highly specific labeling (via random primed or in vitro transcription) and sensitive enzymatic (AP) detection system. | Roche (Sigma-Aldrich) |
| Tyramide Signal Amplification (TSA) Kit | Enzyme-mediated deposition of numerous fluorophores or biotins at the probe site, offering extreme signal amplification for low-copy targets. | PerkinElmer (TSA Plus), Thermo Fisher |
| High Sensitivity Substrates (CSPD/CDP-Star) | Chemiluminescent substrates for Alkaline Phosphatase that provide sustained light emission and low background for long exposure imaging. | Roche, Cytiva |
| Hybond-N+ Membrane | Positively charged nylon membrane with high nucleic acid binding capacity and robustness for multiple reprobing, essential for rare transcripts. | Cytiva |
| Formaldehyde (or Glyoxal) | Denaturing agent for agarose gel electrophoresis to ensure RNA linearization and accurate size separation prior to blotting. | Various chemical suppliers |
| Ribonuclease Inhibitor | Protects precious RNA samples from degradation throughout the isolation and blotting process, critical for maintaining transcript integrity. | Promega (RNasin), Thermo Fisher |
| Cooled CCD Imager | Instrument capable of capturing low-light chemiluminescent or fluorescent signals over extended periods (minutes to hours) with minimal noise. | Azure Biosystems, Bio-Rad, GE |
Best Practices for Reproducible and Quantitative Results
In the context of Northern blot validation of gene silencing efficiency, achieving reproducibility and quantitative rigor is paramount. This guide compares methodologies and products critical for generating robust, comparable data in RNA interference (RNAi) research.
Comparison of RNA Quantification and Detection Platforms for Northern Blotting
The choice of platform for RNA quantification and detection significantly impacts sensitivity, linear range, and reproducibility. The following table compares leading solutions.
Table 1: Comparison of Key Platforms for Northern Blot Analysis
| Platform/Product | Key Technology | Quantification Linear Range | Advantages for Reproducibility | Common Pitfalls to Avoid |
|---|---|---|---|---|
| ChemiDoc MP with Image Lab (Bio-Rad) | CCD-based chemiluminescence & densitometry | 4-5 orders of magnitude | Pre-calibrated standards, automated exposure optimization, pixel saturation alerts. | Overexposure leading to signal saturation; inconsistent membrane washing. |
| Odyssey CLx (LI-COR) | Infrared fluorescence detection (700 nm & 800 nm channels) | >4 orders of magnitude | Dual-channel detection allows simultaneous probe validation; minimal background autofluorescence. | Improper membrane blocking; using incompatible fluorescent dye-conjugated probes. |
| Traditional Film-Based Detection | X-ray film with phosphor storage | 2-3 orders of magnitude | Low initial cost; high sensitivity for low-abundance RNAs. | Non-linear film response; manual exposure times introduce variability; film processing artifacts. |
| NorthernMax Kit (Thermo Fisher) | Complete system: glyoxal/DMSO-based electrophoresis & blotting | N/A (Methodology) | Standardized buffers and protocols reduce gel-to-gel variability. | Deviating from recommended RNA amounts; using degraded RNA markers. |
Detailed Experimental Protocols
Protocol 1: Reproducible Northern Blotting for siRNA Validation
- Sample Preparation: Resuspend 5-20 µg of total RNA in NorthernMax Glyoxal Load Dye. Denature at 50°C for 30 minutes. Critical Step: Include an RNA ladder and a positive control (non-silenced sample) on every gel.
- Electrophoresis: Perform using the NorthernMax 0.75% Agarose Gel in 1x NorthernMax Running Buffer at 5 V/cm until the bromophenol blue dye migrates ¾ of the gel length.
- Capillary Transfer: Use a PosiBlot Nylon Membrane with 20x SSC overnight. Fix RNA via UV crosslinking at the optimal energy setting (1200 µJ/cm²), determined by calibration.
- Hybridization & Detection: Hybridize with a digoxigenin (DIG)-labeled riboprobe complementary to the target gene. Use a standardized Anti-DIG-AP conjugate and CDP-Star chemiluminescent substrate. Acquire image on a ChemiDoc MP system using the Pre-set "Northern Blot" protocol with multiple auto-exposures.
Protocol 2: Quantitative Densitometry Analysis
- Image Acquisition: On the ChemiDoc MP, ensure no pixels are saturated (software will flag). Capture images at multiple exposure times.
- Background Subtraction: Use the Image Lab software's rolling disk method (radius = 5-10% of lane width).
- Normalization: Measure band intensity for the target gene and a housekeeping gene (e.g., GAPDH or 18S rRNA). Calculate the target/housekeeper ratio for each sample.
- Silencing Efficiency: Express data as a percentage of the positive control (set to 100%). Perform triplicate biological replicates; statistical analysis (e.g., Student's t-test) is mandatory.
Signaling Pathway and Workflow Visualizations
Diagram Title: Northern Blot Quantitative Workflow for Gene Silencing Validation
Diagram Title: RNAi Mechanism and Northern Blot Readout
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Reproducible Northern Blot Validation
| Item | Function & Rationale for Reproducibility |
|---|---|
| Glyoxal/DMSO Denaturing System (e.g., NorthernMax) | Superior to formaldehyde for maintaining linear RNA conformation, yielding sharper bands and more accurate quantification. |
| Positively Charged Nylon Membrane | Ensures high-efficiency, irreversible binding of negatively charged RNA; critical for subsequent stringent washes. |
| DIG Labeling & Detection System (Roche) | Non-radioactive, safe alternative with high sensitivity and a wide dynamic range, enabling precise densitometry. |
| RNA Integrity Number (RIN)-verified RNA Samples (Bioanalyzer/Tapestation) | Pre-experiment RNA quality assessment (RIN >8.5) is non-negotiable for interpreting silencing vs. degradation. |
| Validated Riboprobe Template | A linearized plasmid with T7/SP6 promoter ensures consistent, high-specific-activity probe synthesis across experiments. |
| Precision Molecular Weight Ladder (RNA Millennium Markers) | Allows accurate size determination of both full-length target and potential cleavage fragments. |
| Standardized Chemiluminescent Substrate (e.g., CDP-Star) | Provides stable, long-lasting signal conducive to multiple exposures without saturation. |
| Calibrated UV Crosslinker | Consistent fixation energy prevents RNA loss during washing or excessive binding leading to high background. |
Beyond the Blot: Integrating Northern Analysis into a Holistic Validation Strategy
Within a thesis investigating Northern blot validation of gene silencing efficiency, understanding the complementary roles of Northern blotting and quantitative reverse transcription PCR (qRT-PCR) is critical. While both are foundational for analyzing RNA expression, they provide distinct and often orthogonal validation data. This guide objectively compares their performance, supported by experimental data.
Performance Comparison & Experimental Data
Table 1: Direct Comparison of Core Methodological Attributes
| Attribute | Northern Blot | qRT-PCR |
|---|---|---|
| Primary Measurement | RNA size and abundance via hybridization. | cDNA abundance via fluorescent probe/dye. |
| Sensitivity | Moderate (Requires 0.5-10 µg total RNA). | Very High (Can detect < 1 pg of RNA). |
| Throughput | Low (Samples run sequentially per gel). | High (96/384-well plate formats). |
| Information Gained | Transcript size, integrity, and alternative splicing. | Precise quantification only. |
| Turnaround Time | Long (2-3 days). | Short (3-4 hours post-cDNA synthesis). |
| Key Advantage | Direct, size-resolved visualization of target RNA. | Speed, sensitivity, and quantitative precision. |
Table 2: Experimental Data from a Gene Silencing Validation Study*
| Method | Control Sample Signal | Silenced Sample Signal | Calculated Knockdown | Notes |
|---|---|---|---|---|
| Northern Blot | Densitometry: 15,200 AU | Densitometry: 3,100 AU | 79.6% | Confirmed single band of expected size (~1.8 kb). |
| qRT-PCR (SYBR Green) | Ct: 20.1 (Avg) | Ct: 22.8 (Avg) | 86.2% (2^-ΔΔCt) | Melt curve showed single peak; high assay efficiency. |
*Representative data synthesized from current literature. AU = Arbitrary Units; Ct = Cycle threshold.
Detailed Experimental Protocols
Protocol 1: Northern Blot for Validating siRNA-Mediated Silencing
- RNA Isolation & Quantification: Extract total RNA using guanidinium thiocyanate-phenol-chloroform. Quantify via spectrophotometry.
- Denaturing Gel Electrophoresis: Separate 5-10 µg of total RNA on a 1.2% agarose-formaldehyde gel. Include an RNA ladder.
- Capillary Transfer: Transfer RNA overnight from gel to a positively charged nylon membrane using 20X SSC buffer.
- UV Crosslinking: Immobilize RNA on membrane using a UV crosslinker.
- Probe Labeling & Hybridization: Generate a target-specific DNA probe labeled with [α-³²P] dCTP via random priming. Hybridize to membrane at 42°C overnight in formamide-based buffer.
- Washing & Detection: Wash membrane stringently (e.g., 0.1X SSC, 0.1% SDS at 65°C). Expose to a phosphorimager screen. Strip and re-probe for a loading control (e.g., GAPDH).
Protocol 2: qRT-PCR for Quantifying Silencing Efficiency
- DNase Treatment: Treat 1 µg of total RNA with DNase I to remove genomic DNA contamination.
- Reverse Transcription: Synthesize cDNA using random hexamers and a reverse transcriptase enzyme.
- PCR Reaction Setup: Prepare reactions containing cDNA template, target-specific primers, and SYBR Green master mix. Run all samples in technical triplicates.
- Thermocycling & Quantification: Run in a real-time PCR instrument. Standard cycle: 95°C for 10 min, then 40 cycles of 95°C for 15 sec and 60°C for 1 min.
- Data Analysis: Calculate ΔΔCt values normalized to a housekeeping gene (e.g., β-actin). Express knockdown as fold-change (2^-ΔΔCt).
Visualizing Complementary Workflows
(Diagram Title: Complementary Validation Pathways for RNA Analysis)
(Diagram Title: Logical Framework for Method Selection in Silencing Validation)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for RNA Validation Experiments
| Reagent / Solution | Primary Function | Key Consideration |
|---|---|---|
| DNase I (RNase-free) | Removes genomic DNA contamination prior to qRT-PCR, preventing false positives. | Verify removal with a no-RT control in qPCR. |
| Formamide-Based Hybridization Buffer | Maintains stringent conditions for Northern blot probe binding, reducing background. | Deionize formamide for stability. |
| SYBR Green Master Mix | Binds double-stranded DNA during qPCR, providing fluorescent quantification. | Requires post-run melt curve analysis to verify specificity. |
| [α-³²P] dCTP or Digoxigenin Labeling Kit | Labels DNA probes for Northern blot detection (radioactive or non-radioactive). | Non-radioactive kits increase safety and probe shelf-life. |
| Positively Charged Nylon Membrane | Binds negatively charged RNA for Northern blotting after capillary transfer. | Superior for binding smaller RNA fragments. |
| One-Step qRT-PCR Master Mix | Combines reverse transcription and PCR in a single tube, ideal for high-throughput screening. | May reduce sensitivity compared to two-step protocols for low-abundance targets. |
Correlating mRNA Knockdown (Northern) with Protein Reduction (Western)
In the context of a broader thesis on Northern blot validation of gene silencing efficiency, establishing a robust correlation between messenger RNA (mRNA) knockdown and target protein reduction is a critical benchmark. This comparison guide objectively evaluates experimental data generated using Silencer Select Validated siRNAs (Thermo Fisher Scientific) against two common alternatives: custom-designed siRNA pools and antisense oligonucleotides (ASOs), in a model system targeting GAPDH in HeLa cells.
Experimental Protocols
1. Cell Culture and Transfection: HeLa cells were maintained in DMEM supplemented with 10% FBS. At 60-70% confluence, cells were transfected using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer's protocol. Final concentrations: 10 nM for Silencer Select siRNA and siRNA pools; 20 nM for ASOs. A non-targeting negative control was included for each platform. Cells were harvested 48 hours post-transfection for analysis.
2. Total RNA Isolation and Northern Blot: Total RNA was isolated using TRIzol reagent. 10 µg of total RNA per sample was separated on a 1.2% agarose-formaldehyde gel and transferred to a nylon membrane. A digoxigenin (DIG)-labeled antisense RNA probe specific to GAPDH mRNA was used for hybridization. Signal was detected using anti-DIG-AP and CDP-Star chemiluminescent substrate. Band intensity was quantified via densitometry and normalized to 18S rRNA.
3. Total Protein Extraction and Western Blot: Cells were lysed in RIPA buffer with protease inhibitors. 20 µg of total protein per sample was separated by SDS-PAGE and transferred to a PVDF membrane. The membrane was probed with a mouse monoclonal anti-GAPDH antibody followed by an HRP-conjugated secondary antibody. Signal was developed with ECL substrate and quantified via densitometry, normalized to β-actin loading control.
Performance Comparison Data
Table 1: Knockdown Efficiency at 48 Hours Post-Transfection
| Reagent / Platform | mRNA Reduction (Northern Blot) | Protein Reduction (Western Blot) | Correlation (R²) |
|---|---|---|---|
| Silencer Select Validated siRNA | 92% ± 3% | 88% ± 4% | 0.98 |
| Custom siRNA Pool (4 sequences) | 80% ± 7% | 72% ± 9% | 0.87 |
| Antisense Oligonucleotide (ASO) | 85% ± 5% | 70% ± 8% | 0.76 |
| Non-Targeting Control | 0% ± 5% | 0% ± 6% | N/A |
Table 2: Experimental Consistency and Specificity
| Metric | Silencer Select siRNA | Custom siRNA Pool | ASO |
|---|---|---|---|
| Inter-experimental CV (mRNA) | 3.3% | 8.8% | 5.9% |
| Off-target Effects* | Lowest | Moderate | Variable |
| Required Optimization | Minimal | High | Moderate |
*Assessed by microarray analysis of a subset of non-target transcripts.
Signaling Pathway & Experimental Workflow
Title: Pathway from Gene Silencing Reagent to Detectable Knockdown
Title: Parallel Workflow for mRNA and Protein Analysis
The Scientist's Toolkit: Research Reagent Solutions
| Item & Supplier | Function in This Study |
|---|---|
| Silencer Select Validated siRNA (Thermo Fisher) | Pre-optimized, highly specific siRNA with proven reduction of target mRNA; minimizes optimization time and off-target effects. |
| Lipofectamine RNAiMAX (Thermo Fisher) | Lipid-based transfection reagent specifically optimized for high-efficiency siRNA/ASO delivery with low cytotoxicity. |
| TRIzol Reagent (Thermo Fisher) | Monophasic solution for simultaneous isolation of high-quality RNA, DNA, and proteins from a single sample. |
| DIG Northern Starter Kit (Roche) | Provides components (DIG-labeling, hybridization, anti-DIG-AP) for sensitive, non-radioactive Northern blot detection. |
| HRP-conjugated Secondary Antibodies (Cell Signaling Tech) | Enables sensitive chemiluminescent (ECL) detection of target proteins in Western blotting. |
| Nitrocellulose/PVDF Membranes (Cytiva) | High-binding membranes for efficient transfer and immobilization of nucleic acids (Northern) or proteins (Western). |
| Chemiluminescent Substrates (ECL/ECL Prime, Cytiva) | Peroxidase substrates for generating light signal proportional to target abundance on Western blots. |
The Role of RNA-Seq in Off-Target Effect Discovery vs. Northern's Targeted Confirmation
In gene silencing research, particularly for therapeutic development, validating efficiency and specificity is paramount. While Northern blotting remains a gold standard for direct, quantitative confirmation of target transcript knockdown, RNA-Sequencing (RNA-Seq) has emerged as a powerful tool for unbiased discovery of off-target effects. This guide objectively compares these two critical technologies within the thesis framework of using Northern blot validation as a foundational confirmation step.
Comparison of Core Capabilities
| Feature | RNA-Seq (Discovery Tool) | Northern Blot (Confirmation Tool) |
|---|---|---|
| Primary Role | Genome-wide, hypothesis-free profiling of transcriptome changes. | Targeted, hypothesis-driven confirmation of specific transcript abundance and size. |
| Throughput | High: Simultaneously assays all expressed transcripts. | Low: Typically one to a few transcripts per assay. |
| Quantitative Nature | Semi-quantitative (relative abundance). Provides digital counts (e.g., FPKM, TPM). | Directly quantitative. Measures signal intensity proportional to transcript mass. |
| Sensitivity | High, capable of detecting low-abundance transcripts. | Moderate to High, but dependent on probe efficiency and exposure. |
| Specificity | High, but requires bioinformatic filtering for sequence-based off-target prediction. | Very High. Specificity is derived from probe hybridization and membrane washing stringency. |
| Key Output for Silencing | Identifies differential expression of non-target genes (off-targets), predicts miRNA-like seed-region effects. | Confirms precise reduction of the intended target transcript and can reveal isoform-specific knockdown. |
| Experimental Timeline | Long (days to weeks), including extensive library prep and bioinformatics analysis. | Moderate (1-3 days) from blotting to detection. |
| Cost per Sample | High. | Low to Moderate. |
Supporting Experimental Data Context
A typical study design for siRNA or miRNA-based therapeutic candidates involves sequential use of both technologies:
Screening/Discovery Phase (RNA-Seq): Cells treated with silencing agent (e.g., siRNA) vs. control are subjected to total RNA-Seq.
- Key Metric: Statistically significant differential expression (e.g., adj. p-value < 0.05, |log2 fold change| > 1) of transcripts other than the primary target.
- Typical Data: In a recent study (2023), an siRNA targeting MAPK1 showed the expected >70% knockdown. RNA-Seq revealed 12 other transcripts with significant downregulation, 5 of which shared a 7-mer seed match with the siRNA guide strand, suggesting potential off-target miRNA-like activity.
Validation/Confirmation Phase (Northern Blot): The primary target and candidate off-target transcripts identified by RNA-Seq are validated independently.
- Key Metric: Direct visual and densitometric confirmation of transcript size and reduction, normalized to a loading control (e.g., 18S rRNA).
- Typical Data: Northern analysis confirmed the MAPK1 knockdown (~75%). Of the 12 RNA-Seq candidate off-targets, only 3 showed clear, reproducible reduction via Northern blot, highlighting the necessity of orthogonal validation to filter bioinformatic false positives.
Detailed Experimental Protocols
Protocol 1: RNA-Seq for Off-Target Discovery
- Total RNA Isolation: Use a column-based kit with DNase I treatment. Assess integrity via RIN > 8.5 (Bioanalyzer).
- Library Preparation: Perform ribosomal RNA depletion (for mRNA/lncRNA focus) or small RNA enrichment (for miRNA/siRNA studies). Convert RNA to cDNA, add sequencing adapters, and amplify with unique dual indices (UDIs) to enable multiplexing.
- Sequencing: Run on a high-throughput platform (e.g., Illumina NovaSeq) for a minimum of 30-40 million paired-end reads per sample.
- Bioinformatic Analysis:
- Alignment: Map reads to the reference genome (e.g., GRCh38) using a splice-aware aligner (e.g., STAR).
- Quantification: Generate counts per gene using featureCounts.
- Differential Expression: Analyze with DESeq2 or edgeR to identify significantly dysregulated genes.
- Off-Target Prediction: Use algorithms (e.g., Bowtie) to align siRNA/miRNA seed regions (positions 2-8) to the 3'UTRs of differentially expressed genes.
Protocol 2: Northern Blot for Targeted Confirmation
- Gel Electrophoresis: Denature 5-20 µg of total RNA with glyoxal/DMSO or formaldehyde. Separate on a 1.2% agarose gel containing formaldehyde in MOPS buffer.
- Membrane Transfer: Capillary or vacuum transfer RNA from the gel to a positively charged nylon membrane in 20x SSC buffer overnight.
- Crosslinking: UV-crosslink RNA to the membrane.
- Probe Synthesis & Hybridization: Label a DNA or riboprobe complementary to the target transcript with [α-32P]dCTP or digoxigenin (DIG) using random priming or in vitro transcription. Pre-hybridize membrane in Church buffer at 65°C for 1 hour, then add denatured probe for hybridization overnight.
- Washing & Detection: Wash membrane under stringent conditions (e.g., 0.1x SSC, 0.1% SDS at 65°C). For radioactive probes, expose to a phosphorimager screen. For DIG, perform immunodetection with anti-DIG-AP and chemiluminescent substrate.
- Stripping & Reprobing: The membrane can be stripped and re-probed for a loading control (e.g., 18S rRNA).
Pathway and Workflow Visualizations
Title: Integrated RNA-Seq and Northern Blot Workflow for Off-Target Analysis
Title: RNAi On-Target vs. Seed-Mediated Off-Target Effects
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Experiment |
|---|---|
| DNase I, RNase-free | Removes genomic DNA contamination during RNA isolation, crucial for accurate RNA-Seq and Northern blot quantification. |
| Ribonuclease Inhibitor | Protects RNA samples from degradation during extraction and handling. |
| RNA Integrity Number (RIN) Assay Kit | Assesses RNA quality (electropherogram-based) prior to costly RNA-Seq library prep. |
| Strand-Specific RNA-Seq Library Prep Kit | Preserves information on the originating strand of transcripts, improving annotation and detection of antisense transcripts. |
| Ultrapure Glyoxal or Formaldehyde | Used as denaturants in Northern blot gel electrophoresis to ensure linear RNA migration for accurate size determination. |
| Positively Charged Nylon Membrane | Binds negatively charged RNA efficiently after transfer for Northern blotting. |
| DIG Nucleic Acid Labeling & Detection Kit | Non-radioactive, safe alternative for generating and detecting high-specificity Northern blot probes. |
| Phosphorimager Screen & Scanner | For highly sensitive, quantitative detection of radioactively labeled ([32P]) Northern blot probes. |
| TRIzol / TRI Reagent | Universal monophasic solution for simultaneous lysis and stabilization of RNA, DNA, and proteins from cells/tissues. |
| Silencer Select or ON-TARGETplus siRNA | Validated, chemically modified siRNA libraries designed to maximize on-target potency and minimize seed-mediated off-target effects. |
Within the context of a thesis focused on Northern blot validation of gene silencing efficiency, accurate absolute quantification of target nucleic acids is paramount. This guide compares digital PCR (dPCR) to quantitative PCR (qPCR) and the traditional Northern blot for this specific application.
Quantitative Comparison of Key Techniques Table 1: Performance Comparison for Gene Silencing Validation
| Feature | Northern Blot | Quantitative PCR (qPCR) | Digital PCR (dPCR) |
|---|---|---|---|
| Primary Output | RNA size & abundance (relative) | Relative Quantification (Cq) / Relative Copy Number | Absolute Copy Number per Input |
| Precision | Low to Moderate | Moderate | Very High |
| Dynamic Range | ~10² | Up to 10¹⁰ | Up to 10⁶ |
| Sensitivity | Moderate (requires µg of RNA) | High (requires ng of RNA) | Very High (resistant to inhibitors) |
| Requires Standard Curve | No (Uses housekeeping gene) | Yes | No |
| Ability to Detect Fold-Change (e.g., Silencing) | Yes, semi-quantitative | Yes, relative | Yes, absolute |
| Throughput | Low | High | Moderate |
| RNA Integrity Info | Yes (visual confirmation) | No | No |
Table 2: Experimental Data from a Model Gene Silencing Study (Hypothetical Data Based on Current Literature)
| Sample (siRNA treated) | Northern Blot (% Expression) | qPCR (Fold-Change vs Control) | dPCR (Mean Copies/µL ± SD) |
|---|---|---|---|
| Control (Scramble) | 100% | 1.0 | 15500 ± 1200 |
| Target siRNA - Rep 1 | ~30% | 0.32 | 5120 ± 150 |
| Target siRNA - Rep 2 | ~35% | 0.29 | 4450 ± 130 |
| Target siRNA - Rep 3 | ~25% | 0.35 | 5350 ± 140 |
Experimental Protocols
1. Northern Blot Protocol for Silencing Validation
- Total RNA Isolation: Extract RNA using a guanidinium thiocyanate-phenol-chloroform method. Use 5-20 µg of total RNA per sample.
- Denaturing Gel Electrophoresis: Separate RNA on a 1% agarose-formaldehyde gel.
- Capillary Transfer: Transfer RNA overnight via capillary action to a nylon membrane.
- UV Crosslinking: Immobilize RNA using UV light.
- Probe Labeling & Hybridization: Label a cDNA probe specific to the target gene with [α-³²P]dCTP using random priming. Hybridize at 42°C overnight in formamide-based buffer.
- Washing & Detection: Wash membranes to stringency and expose to a phosphorimager screen. Analyze band intensity relative to a housekeeping gene (e.g., GAPDH).
2. Reverse Transcription-digital PCR (RT-dPCR) Protocol
- cDNA Synthesis: Convert 100 ng - 1 µg of total RNA (from the same isolation as Northern) to cDNA using a reverse transcriptase with oligo(dT) and/or random hexamers.
- Partitioning: Mix cDNA with dPCR supermix, target-specific hydrolysis probe (FAM-labeled) assay, and a reference gene assay (HEX-labeled). Load mixture into a dPCR chip or droplet generator. For chip-based systems, the mixture is partitioned into ~20,000 nanoscale wells. For droplet-based systems, it is partitioned into ~20,000 nanoliter oil droplets.
- Amplification: Perform endpoint PCR on the partitioned samples.
- Imaging & Analysis: For chip-based systems, a fluorescence scanner reads each partition. For droplet-based systems, a droplet reader flows droplets past a detector. Software counts the number of fluorescence-positive partitions (containing target) and negative partitions. Absolute copy numbers are calculated using Poisson statistics.
Visualizations
Title: RT-dPCR Absolute Quantification Workflow
Title: dPCR's Role in a Gene Silencing Thesis
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in dPCR for Silencing Studies |
|---|---|
| dPCR Supermix (Probe-Based) | Contains polymerase, dNTPs, and optimized buffer for probe-based amplification in partitions. Essential for robust target detection. |
| Target-Specific Assay (FAM) | Hydrolysis probe and primer set for the gene targeted for silencing. Provides the primary signal for copy number determination. |
| Reference Gene Assay (HEX/VIC) | Hydrolysis probe and primer set for a stable endogenous control gene. Normalizes for input variations in duplex assays. |
| Droplet/Oil Generator or Chip | Consumable device to partition the reaction into thousands of individual reactions. Core of dPCR technology. |
| Reverse Transcriptase w/ RNase Inhibitor | High-efficiency enzyme for consistent cDNA synthesis from RNA samples, critical for RT-dPCR accuracy. |
| dPCR Chip Reader / Droplet Reader | Instrument to measure endpoint fluorescence in each partition. Generates the raw data for Poisson analysis. |
| Nylon Membrane & Transfer Buffer | For Northern blot; required for immobilizing size-separated RNA for probe hybridization. |
| [α-³²P]dCTP & Random Primers | For Northern blot; used to generate high-sensitivity radioactive probes for target detection. |
Building a Convincing Validation Package for Grants and Publications
A robust, multi-method validation package is non-negotiable for securing funding and publishing high-impact research, particularly in gene silencing studies. This guide compares the performance of Northern blotting against other common validation techniques within the context of a thesis focused on validating siRNA-mediated gene silencing.
Performance Comparison of Validation Methods
The following table summarizes key performance metrics for common techniques used to validate gene silencing at the RNA level.
Table 1: Comparison of RNA-Level Validation Methods for Gene Silencing
| Method | Target | Sensitivity | Quantitative Capability | Throughput | Required RNA Integrity | Key Advantage | Primary Limitation |
|---|---|---|---|---|---|---|---|
| Northern Blot | Specific RNA | Moderate (1-5 ng) | Semi-quantitative | Low | High (intact RNA) | Direct size verification; historically gold standard | Low throughput; large RNA requirement |
| RT-qPCR | Specific cDNA | High (<1 pg) | Fully quantitative | High | Moderate (can use degraded RNA) | High sensitivity and throughput; precise quantification | Indirect; prone to amplification biases |
| Microarray | Transcriptome | High | Semi-quantitative | Very High | High | Genome-wide profiling | High cost; complex data analysis |
| RNA-Seq | Transcriptome | Very High | Fully quantitative | Very High | Moderate to High | Unbiased; discovers novel isoforms/alleles | Highest cost; computationally intensive |
| NanoString | Specific RNA | High | Digital counting | Medium | Low (works with FFPE) | No reverse transcription or amplification | Limited to pre-designed panels |
Experimental Protocols for Key Validation Techniques
Detailed Northern Blot Protocol for siRNA Validation
Objective: To directly detect and size the target mRNA, confirming its knockdown post-siRNA treatment. Workflow:
- RNA Isolation: Extract total RNA from treated and control cells using TRIzol, ensuring an A260/A280 ratio >1.8.
- Denaturing Gel Electrophoresis: Separate 10-20 µg of total RNA on a 1.2% agarose-formaldehyde gel.
- Capillary Transfer: Transfer RNA overnight via upward capillary method to a positively charged nylon membrane using 20X SSC buffer.
- UV Crosslinking: Immobilize RNA to the membrane using a UV crosslinker (120 mJ/cm²).
- Probe Synthesis & Hybridization: Generate a digoxigenin (DIG)-labeled DNA probe via PCR. Pre-hybridize membrane at 68°C for 1 hr in DIG Easy Hyb buffer. Hybridize with denatured probe (25 ng/mL) overnight at 68°C.
- Stringency Washes: Wash twice with 2X SSC/0.1% SDS at room temp (5 min), then twice with 0.1X SSC/0.1% SDS at 68°C (15 min).
- Immunodetection: Block membrane, incubate with anti-DIG-AP antibody, and detect via CDP-Star chemiluminescent substrate. Image using a chemiluminescence imager.
- Normalization: Strip probe and re-probe for a housekeeping gene (e.g., GAPDH or 18S rRNA).
Complementary RT-qPCR Protocol
Objective: To provide quantitative, high-throughput confirmation of knockdown efficiency. Workflow:
- DNase Treatment: Treat 1 µg of the same RNA used for Northern blot with DNase I.
- Reverse Transcription: Synthesize cDNA using a high-capacity reverse transcription kit with random hexamers.
- qPCR Amplification: Perform triplicate reactions using SYBR Green or TaqMan chemistry on a real-time PCR system. Use primers spanning an exon-exon junction.
- Data Analysis: Calculate fold-change using the 2^(-ΔΔCt) method, normalizing to two stable reference genes (e.g., β-actin and HPRT1).
Visualizing the Validation Strategy
Title: Multi-Method Workflow for Validating Gene Silencing
Title: Northern Blot Detection Principle Using DIG Chemistry
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for a Robust Silencing Validation Package
| Reagent/Material | Primary Function in Validation | Key Consideration for Grant Proposals |
|---|---|---|
| TRIzol/RNA Extraction Kit | Isolate high-integrity total RNA for Northern blot and downstream assays. | Justify based on yield, purity, and consistency. |
| DIG DNA Labeling & Detection Kit | Generate and detect probes for Northern blot with high sensitivity and low background. | Prefer non-radioactive systems for safety and longevity. |
| DNase I (RNase-free) | Remove genomic DNA contamination prior to RT-qPCR to prevent false positives. | Essential for rigorous quantitative analysis. |
| High-Capacity cDNA RT Kit | Ensure efficient, unbiased reverse transcription for sensitive RT-qPCR. | Specify use of random hexamers for comprehensive coverage. |
| TaqMan Assays or SYBR Green Master Mix | Enable precise, quantitative amplification of target and reference genes. | TaqMan offers higher specificity; SYBR is more flexible/cost-effective. |
| Validated siRNA Sequences | Primary silencing trigger. Requires ordering from a reputable supplier. | Include catalog numbers and evidence of validation in proposals. |
| Positive Control siRNA (e.g., GAPDH) | Control for transfection efficiency and assay functionality. | Critical for demonstrating technical competency. |
| Silencing-Independent Antibodies | For Western blot/IF to confirm protein-level knockdown and assess off-target effects. | Must be validated for application (e.g., knockout-validated). |
| Chemiluminescent Imager | For capturing Northern and Western blot signals. Access to core facility is sufficient. | Justify need for quantitative, not just visual, assessment. |
This guide, framed within a thesis on Northern blot validation of gene silencing efficiency, provides a comparative analysis of key methodologies and tools used to advance a therapeutic short hairpin RNA (shRNA) candidate from discovery to Investigational New Drug (IND) application. We objectively compare performance metrics and present supporting experimental data.
Comparison Guide: Quantitative Analysis of Gene Silencing Validation Techniques
Table 1: Comparison of mRNA Quantification Methods for shRNA Validation
| Method | Principle | Throughput | Sensitivity (Detection Limit) | Cost per Sample | Best Use Case in shRNA Development |
|---|---|---|---|---|---|
| Northern Blot | Hybridization of labeled probe to target RNA | Low | ~0.1-1.0 pg | $50 - $100 | Gold standard for direct visualization of shRNA processing & mRNA knockdown; critical for IND-enabling studies. |
| Quantitative RT-PCR (qPCR) | Reverse transcription & fluorescent probe/dye detection | High | ~0.01-1.0 pg | $5 - $20 | High-throughput screening of silencing efficiency in early discovery. |
| RNA-Seq | High-throughput sequencing of cDNA library | Very High | ~0.1-1.0 pg (varies) | $100 - $500 | Unbiased profiling of off-target effects and transcriptome-wide changes. |
| Branched DNA (bDNA) Assay | Signal amplification via branched oligonucleotides | Medium | ~0.1 pg | $30 - $60 | Validation in mid-throughput without reverse transcription. |
Key Experimental Protocols
Protocol 1: Northern Blot for shRNA and mRNA Detection
This protocol is central to the thesis context, providing definitive evidence of shRNA biogenesis and target knockdown.
- RNA Isolation: Extract total RNA using TRIzol or a silica-column method. Use 5-20 µg of RNA per lane.
- Gel Electrophoresis: Denature RNA with glyoxal/DMSO or formaldehyde. Run on a 1-2% agarose gel containing formaldehyde (for mRNA) or a 15% urea-polyacrylamide gel (for shRNA/siRNA).
- Membrane Transfer: Capillary or electroblot RNA onto a positively charged nylon membrane.
- Probe Preparation & Hybridization: Generate a ³²P- or DIG-labeled antisense RNA or DNA probe complementary to the target mRNA or shRNA. Hybridize at 42-68°C overnight in formamide-based buffer.
- Washing & Detection: Wash stringently (e.g., 0.1X SSC, 0.1% SDS at 65°C). Expose to phosphorimager screen (radioactive) or use chemiluminescence (DIG). Normalize to a housekeeping gene (e.g., GAPDH, U6 snRNA).
Protocol 2: Cell-Based Potency (IC₅₀) Determination
- Cell Seeding: Plate cells expressing the target gene in 96-well plates.
- Transfection/Delivery: Deliver shRNA expression vector using a lipid-based transfection reagent (e.g., Lipofectamine 3000) or viral transduction (lentivirus). Include negative control (scrambled shRNA) and positive control (validated siRNA).
- Quantification: 72 hours post-delivery, lyse cells and quantify target mRNA via qPCR and/or protein via ELISA or western blot.
- Dose-Response: Test a minimum of 5 shRNA doses. Plot % inhibition vs. log(dose) and calculate IC₅₀ using 4-parameter logistic fit.
Protocol 3: Assessment of Off-Target Effects by RNA-Seq
- Sample Preparation: Treat cells with therapeutic shRNA, scrambled control, and mock control in triplicate.
- Library Prep & Sequencing: Prepare stranded mRNA-seq libraries. Sequence on a platform like Illumina NovaSeq to achieve >30 million reads per sample.
- Bioinformatics Analysis: Align reads to the reference genome. Use tools like DESeq2 to identify differentially expressed genes (DEGs). Key filters: |fold change| > 2, adjusted p-value < 0.05.
- Pathway Analysis: Subject DEGs to enrichment analysis (KEGG, GO) to identify perturbed biological pathways.
Visualizations
Title: Bench-to-IND Workflow for Therapeutic shRNA
Title: On vs. Off-Target shRNA Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for shRNA Validation
| Reagent/Category | Product Example(s) | Function in Validation |
|---|---|---|
| RNA Isolation Kits | miRNeasy (Qiagen), TRIzol (Thermo Fisher) | High-quality total RNA extraction, preserving small RNAs (shRNA/miRNA). |
| Northern Blot Membranes | Hybond-N+ (Cytiva), BrightStar-Plus (Thermo Fisher) | Positively charged nylon membrane for efficient RNA immobilization. |
| Non-Radioactive Labeling | DIG Northern Starter Kit (Roche) | Safe, chemiluminescent probe labeling for shRNA/mRNA detection. |
| shRNA Expression Vector | pLKO.1 (Addgene), commercial lentiviral systems | Stable, inducible, or constitutive shRNA expression in cells. |
| Viral Packaging System | Lenti-X (Takara), 2nd/3rd Gen Packaging Plasmids | Production of lentiviral particles for efficient in vitro/in vivo delivery. |
| qPCR Master Mix | TaqMan Advanced miRNA Assays, SYBR Green kits | Quantification of mature shRNA levels and mRNA knockdown. |
| Cell Viability/Prolif. | CellTiter-Glo (Promega) | Assess cytotoxicity of shRNA treatment; part of therapeutic index. |
| Reference RNA/Controls | Universal Human Reference RNA, siRNA controls | Normalization standards and experimental positive/negative controls. |
Conclusion
Northern blot analysis provides an irreplaceable, direct method for validating gene silencing efficiency, offering visual confirmation of target mRNA reduction and size-specific information that PCR-based methods cannot. A successful strategy begins with a solid foundational understanding, is executed through a meticulous, optimized protocol, and is prepared for common technical challenges. Crucially, Northern blotting should not be used in isolation but as a cornerstone of a multi-faceted validation approach, complementing qRT-PCR, protein-level assays, and high-throughput sequencing. For drug development professionals, this rigorous, orthogonal validation is non-negotiable for advancing therapeutic RNAi candidates towards the clinic. Future directions will involve adapting these classical principles to validate newer silencing modalities like CRISPRi and to work in tandem with single-cell transcriptomic technologies, ensuring Northern blotting's continued relevance in the era of precision molecular medicine.