Harnessing Deep Learning for Plant Resistance Gene Prediction: A Comprehensive Guide to PRGminer
This article provides a comprehensive exploration of PRGminer, a state-of-the-art deep learning tool for the high-throughput prediction and classification of plant resistance genes (R-genes).
Harnessing Deep Learning for Plant Resistance Gene Prediction: A Comprehensive Guide to PRGminer
Abstract
This article provides a comprehensive exploration of PRGminer, a state-of-the-art deep learning tool for the high-throughput prediction and classification of plant resistance genes (R-genes). Tailored for researchers, scientists, and professionals in plant science and biotechnology, the content covers foundational knowledge of plant-pathogen interactions and R-gene diversity, delves into the tool's two-phase deep learning architecture and practical application workflow, addresses common implementation challenges and data optimization strategies, and offers a critical evaluation of its performance against traditional methods. By synthesizing the latest research, this guide aims to empower the scientific community to accelerate the discovery of novel R-genes, advancing strategies for breeding disease-resistant crops and enhancing global food security.
Understanding Plant Immunity and the Critical Role of Resistance Genes
Plants have evolved a sophisticated, multi-layered innate immune system to defend against diverse pathogenic threats. This system primarily consists of two interconnected branches: Pattern-Triggered Immunity (PTI) and Effector-Triggered Immunity (ETI). PTI represents the first line of defense, where plant cell surface receptors, known as Pattern Recognition Receptors (PRRs), detect conserved microbial patterns [1]. When this initial defense is breached, the second layer, ETI, is activated through intracellular Nucleotide-binding Leucine-rich Repeat (NLR) receptors that recognize specific pathogen effector proteins, leading to a more potent and specialized immune response [1]. The synergistic interaction between PTI and ETI creates a robust defense network, allowing plants to balance resource allocation between growth and effective immune responses against pathogen colonization [2]. Understanding these mechanisms is crucial for agricultural biotechnology, as it enables the development of disease-resistant crops through advanced breeding techniques and computational tools like PRGminer, a deep learning-based platform for predicting resistance genes [3].
Conceptual Framework of PTI and ETI
Pattern-Triggered Immunity (PTI): The First Layer of Defense
PTI is initiated when PRRs on the plant cell surface recognize Pathogen-Associated Molecular Patterns (PAMPs) or Microbe-Associated Molecular Patterns (MAMPs)—conserved molecules essential for microbial survival [1] [4]. Additionally, plants can detect Damage-Associated Molecular Patterns (DAMPs), which are host-derived molecules released during pathogen invasion [5]. PRRs primarily include Receptor-Like Kinases (RLKs) and Receptor-Like Proteins (RLPs). RLKs contain an extracellular domain for ligand binding, a transmembrane domain, and an intracellular kinase domain, while RLPs have a similar structure but lack the intracellular kinase domain [5].
Key PRRs and their recognized patterns include:
- FLS2 (Flagellin Sensing 2): An LRR-RLK that recognizes bacterial flagellin-derived peptide flg22 [5].
- EFR (EF-Tu Receptor): An LRR-RK that identifies the bacterial elongation factor Tu epitope elf18 [5].
- CERK1 (Chitin Elicitor Receptor Kinase 1): A LysM-containing RK that binds fungal chitin and activates downstream defense signaling [5].
Upon PAMP perception, PRRs often form complexes with co-receptors such as BAK1 (BRI1-Associated Receptor Kinase 1) and SOBIR1 (Suppressor of BIR1-1), initiating a cascade of intracellular signaling events [5]. This signaling involves calcium ion influx, Reactive Oxygen Species (ROS) burst, Mitogen-Activated Protein Kinase (MAPK) activation, and extensive transcriptional reprogramming that collectively establish an anti-microbial environment [1].
Effector-Triggered Immunity (ETI): The Second Layer of Defense
Successful pathogens deliver effector proteins into host cells to suppress PTI and promote virulence [1]. Plants have consequently evolved intracellular NLR receptors that directly or indirectly recognize these effectors, activating ETI [4]. ETI is generally more robust and prolonged than PTI and is often associated with the Hypersensitive Response (HR)—a form of programmed cell death at the infection site that restricts pathogen spread [6] [1].
NLR receptors are categorized based on their N-terminal domains:
- TNLs: Contain Toll/Interleukin-1 Receptor (TIR) domains.
- CNLs: Feature Coiled-Coil (CC) domains [3].
Recent research in Arabidopsis has revealed that ETI activation by TNLs involves two key signaling modules: the EDS1-PAD4-ADR1 module, which is critical for immune priming and growth inhibition, and the EDS1-SAG101-NRG1 module, which primarily mediates HR cell death [6]. The synergistic action of these modules ensures effective pathogen resistance while managing the growth-defense trade-off [6].
PTI-ETI Integration and Synergy
Originally viewed as independent branches, PTI and ETI are now understood to form a unified immune system with synergistic interactions [5]. PTI and ETI activate qualitatively similar defense outputs, including ROS production, calcium influx, and transcriptional reprogramming, suggesting their signaling pathways converge upstream of nuclear events [5]. Studies show that immune components in PTI and ETI have coevolved across plant species, blurring the distinction between the two systems [5]. This synergistic relationship results in a more robust and amplified immune response than either branch could achieve alone.
The following diagram illustrates the core signaling pathways and their convergence in plant immunity.
The Scientist's Toolkit: Research Reagent Solutions
Studying plant immunity requires a diverse array of specialized reagents and tools. The following table catalogues essential research reagents for investigating PTI, ETI, and associated signaling pathways.
Table 1: Key Research Reagents for Plant Immunity Studies
| Reagent Category | Specific Examples | Research Application & Function |
|---|---|---|
| PAMP/MAMP Ligands | flg22, elf18, chitin oligosaccharides (nlp20) | Elicitation of PTI responses; used to study early signaling events (e.g., ROS burst, MAPK activation) and transcriptional reprogramming [5]. |
| Receptor & Co-receptor Constructs | FLS2, EFR, CERK1, BAK1, SOBIR1 (genes, antibodies, mutant lines) | Molecular characterization of PRR complexes; investigating ligand-binding specificity, receptor activation, and downstream signaling [5]. |
| NLR Constructs & Mutants | TNL/CNL expression clones, eds1, pad4, sag101, nrg1 mutant plants | Functional dissection of ETI pathways; defining the roles of specific NLRs and signaling nodes like EDS1 modules in cell death and resistance [6]. |
| Signaling Pathway Reporters | Genetically encoded Ca²⁺ and ROS sensors (e.g., Aequorin, roGFP), MAPK activity antibodies | Real-time monitoring and quantification of key signaling events during both PTI and ETI activation [5] [1]. |
| Hormone Analysis Kits | Salicylic Acid (SA), Jasmonic Acid (JA), Ethylene (ET) quantification kits (ELISA, LC-MS) | Profiling phytohormone levels to understand their crosstalk and role in mediating different immune outputs and systemic resistance [7]. |
| Transcriptional Profiling Tools | Microarrays, RNA-Seq kits, qPCR primers for defense marker genes (e.g., PR1, FRK1) | Global and targeted analysis of gene expression changes during immune responses to identify key regulators and defense pathways [2]. |
Computational Protocol: Predicting R-genes with PRGminer
The identification of resistance (R) genes is a critical step in understanding ETI and advancing plant breeding. PRGminer is a deep learning-based tool designed for high-throughput prediction and classification of R-genes from protein sequences, overcoming limitations of traditional alignment-based methods [3] [8].
PRGminer operates through a structured two-phase prediction system, as illustrated below.
Step-by-Step Prediction Protocol
Input Preparation
- Input Methods: Users can submit protein sequences via three primary methods: 1) entering a valid NCBI or UniProt accession ID; 2) uploading a FASTA file containing single or multiple sequences; or 3) directly pasting FASTA-formatted sequences into the provided text area [9].
- Sequence Requirements: Ensure protein sequences are in standard FASTA format. The tool is designed to handle both full-length and incomplete R-genes, which often pose challenges for automatic annotation pipelines [3].
Execution and Analysis
- Submission: Click the "Run Prediction" button after providing the input. The deep learning model will process the sequences.
- Phase I - Binary Classification: The tool first classifies the input sequence as either an R-gene or a Non-R-gene. This phase uses dipeptide composition features from the protein sequence, achieving an accuracy of 98.75% in k-fold testing and 95.72% on independent datasets [3] [8].
- Phase II - R-gene Classification: Sequences identified as R-genes are further classified into one of eight specific classes. The overall accuracy for this multi-class classification is 97.55% in k-fold testing and 97.21% on independent tests [3] [8].
Output Interpretation
- Results Table: The output includes a table summarizing the Sequence ID, prediction outcome, confidence scores, and detailed classification.
- Confidence Scores: These probabilities indicate the model's certainty in its prediction. A higher score corresponds to greater prediction reliability.
- Download Options: Complete results can be downloaded in CSV, JSON, or FASTA formats, allowing for further offline analysis and integration with other bioinformatics pipelines [9].
Performance Metrics and Validation
PRGminer has been rigorously validated. The high Matthews Correlation Coefficient (MCC) values of 0.98 (training) and 0.91 (independent testing) in Phase I indicate a strong model capable of minimizing false positives and negatives [8]. This performance makes it a valuable tool for accelerating the discovery of novel R-genes in wild plant species and crop relatives, which is fundamental for understanding ETI and developing durable disease resistance.
Experimental Protocol: Dissecting ETI Signaling Modules
This protocol outlines the genetic and transcriptomic approaches to characterize the two major EDS1-dependent signaling modules in ETI, based on methodologies refined in recent research [6].
Experimental Workflow
The following diagram maps the key stages of the experimental process.
Materials and Reagents
- Plant Materials: Wild-type and mutant lines of Arabidopsis thaliana, including:
- Single mutants:
pad4,adr1,sag101,nrg1. - Double mutants:
pad4 adr1,sag101 nrg1. - Higher-order mutants:
eds1null mutant as a control [6].
- Single mutants:
- Pathogen Strain: A bacterial pathogen (e.g., Pseudomonas syringae) expressing an effector protein known to activate TNL-mediated ETI.
- Growth Media: Solid Murashige and Skoog (MS) medium, as required for seedling growth.
- RNA-seq Reagents: Kit for total RNA extraction, reverse transcription kit, library preparation kit, and sequencing platform access.
Step-by-Step Procedure
Plant Growth and Genotyping (Step 1)
- Stratification: Sow Arabidopsis seeds on MS plates and cold-treat at 4°C for 48 hours to synchronize germination.
- Growth: Transfer plates to a controlled environment growth chamber with set conditions (e.g., 22°C, 16h light/8h dark cycle).
- Genotyping: Confirm the genotype of each plant line using PCR-based markers or sequencing before proceeding to experiments.
Pathogen Inoculation and ETI Induction (Step 2)
- Pathogen Culture: Grow the bacterial pathogen in liquid King's B medium overnight with appropriate antibiotics.
- Harvest and Resuspend: Centrifuge the bacterial culture, wash, and resuspend in a suitable buffer (e.g., 10 mM MgCl₂) to a final concentration of 1 × 10^8 CFU/mL for infiltration.
- Infiltrate Leaves: Use a needleless syringe to pressure-infiltrate the bacterial suspension into the abaxial side of leaves from 4-5 week old plants. For negative controls, infiltrate with buffer only (mock inoculation).
Phenotypic Analysis (Step 3)
- Hypersensitive Response (HR) Assay: Visually monitor and document the appearance of localized cell death (tissue collapse and browning) in the infiltrated areas at 24-48 hours post-inoculation (hpi).
- Pathogen Growth Assay:
- At 0 and 3 days post-inoculation (dpi), collect leaf discs from the infiltrated areas.
- Homogenize the discs in buffer, serially dilute the homogenate, and plate on solid medium with antibiotics.
- Count bacterial colonies after a 2-day incubation and calculate the CFU/cm² of leaf tissue.
- Growth Inhibition Measurement: Measure the fresh weight of seedlings or the root length of plants grown on MS plates 10-14 days after pathogen challenge or mock treatment.
Transcriptomic Profiling (Step 4)
- RNA Extraction: At a critical time point (e.g., 6 hpi), harvest leaf tissue from inoculated and mock-treated plants and immediately freeze in liquid nitrogen. Extract total RNA using a commercial kit, including a DNase I digestion step.
- Library Preparation and Sequencing: Assess RNA quality (RIN > 7.0). Prepare RNA-seq libraries from high-quality RNA and sequence on an Illumina platform to generate at least 20 million paired-end reads per sample.
- Bioinformatic Analysis:
- Align sequence reads to the Arabidopsis reference genome.
- Perform differential gene expression analysis to compare pathogen-treated samples to mock controls for each genotype.
- Identify genes that are differentially expressed in wild-type plants but show altered expression in the specific mutants.
Expected Outcomes and Data Analysis
- Phenotypic Data: The
sag101 nrg1mutants are expected to show a strong reduction in HR cell death but retain the ability to limit pathogen growth. Conversely,pad4 adr1mutants will exhibit compromised pathogen growth inhibition but may still display cell death [6]. - Transcriptomic Data: Analysis will reveal that the PAD4-ADR1 module regulates a transcriptional network essential for immune priming and limiting pathogen growth. The SAG101-NRG1 module will control a distinct but overlapping set of genes, more narrowly associated with the execution of cell death [6].
- Validation: Use quantitative RT-PCR to validate the expression of key marker genes identified in the RNA-seq analysis in an independent biological experiment.
The plant immune system is a paradigm of biological complexity, where the layered defenses of PTI and ETI engage in a continuous molecular dance with pathogens. The synergistic relationship between these branches ensures a robust and adaptable response [5]. Cutting-edge research continues to dissect the nuanced signaling modules within ETI, such as the distinct roles of EDS1 complexes, revealing how plants balance effective defense with resource allocation for growth [6]. The integration of traditional genetics with modern computational tools like PRGminer is revolutionizing our ability to identify the key genetic components of this system [3]. This comprehensive understanding, from conceptual frameworks to detailed experimental and computational protocols, provides the foundation for developing next-generation crops with durable, broad-spectrum disease resistance, which is vital for global food security.
Plant disease resistance genes (R-genes) are essential components of the plant immune system, encoding proteins that detect pathogen-derived molecules and initiate robust defense responses [10]. These sophisticated surveillance systems allow plants to recognize invading pathogens and activate signaling cascades that culminate in the production of antimicrobial compounds, reinforcement of cell walls, and in some cases, programmed cell death at infection sites to prevent pathogen spread [3] [11]. The conceptual framework for understanding plant immunity has been organized into a two-branch model comprising pattern-triggered immunity (PTI) and effector-triggered immunity (ETI) [12]. PTI constitutes the first layer of defense, activated when cell-surface receptors recognize conserved pathogen-associated molecular patterns (PAMPs). ETI represents the second, more potent layer, triggered when intracellular receptors detect specific pathogen effector proteins [12] [13].
The genetic basis of these recognition events was first described by the gene-for-gene hypothesis, which posits that for every pathogen avirulence (Avr) gene, there is a corresponding plant R-gene that enables recognition and defense activation [12]. This model has been validated across numerous plant-pathogen systems involving bacteria, fungi, oomycetes, and viruses. Molecular studies have since revealed that R-proteins can be categorized into several major classes based on their structural domains and subcellular localization, with the primary classes being nucleotide-binding leucine-rich repeat receptors (NLRs), receptor-like kinases (RLKs), and receptor-like proteins (RLPs) [13] [14]. Recent genomic analyses of 350 plant species have revealed a surprising concerted expansion and contraction between cell-surface and intracellular immune receptor gene families, suggesting an evolutionary relationship between the two branches of the plant immune system [15].
Structural and Functional Characteristics of Major R-gene Classes
Nucleotide-Binding Leucine-Rich Repeat Receptors (NLRs)
NLRs constitute the largest class of intracellular immune receptors in plants, characterized by a central nucleotide-binding adaptor shared by APAF-1, certain R proteins, and CED-4 (NB-ARC) domain and C-terminal leucine-rich repeats (LRRs) [12] [16]. These proteins function as intracellular immune sensors that detect pathogen effectors directly or indirectly through guard mechanisms that monitor host cellular targets [16] [13]. The N-terminal domain of NLRs is variable and used to classify them into subfamilies: those with Toll/interleukin-1 receptor-like domains (TNLs) and those with coiled-coil domains (CNLs) [10] [13].
Upon pathogen recognition, NLRs undergo conformational changes from ADP-bound (inactive) to ATP-bound (active) states, enabling them to form multiprotein complexes called resistosomes [12] [17]. These resistosomes function as signaling hubs that converge on calcium-permeable channels, triggering downstream immune responses including the hypersensitive response (HR) [17]. The LRR domains facilitate protein-protein interactions and ligand binding, while the NB-ARC domain serves as a molecular switch regulated by nucleotide exchange [10].
Table 1: Major Subclasses of NLR Proteins
| Subclass | N-terminal Domain | Key Structural Features | Representative Examples | Signaling Mechanisms |
|---|---|---|---|---|
| TNL | Toll/Interleukin-1 Receptor (TIR) | TIR-NBS-LRR architecture; TIR domain has NADase activity | RPP4, RPP5, SNC1 [16] | TIR domain generates signaling molecules; Requires EDS1/PAD4 signaling partners [12] |
| CNL | Coiled-Coil (CC) | CC-NBS-LRR architecture; CC domain may form pore structures | RPS2, RPM1, Rpi-blb2 [13] [14] | CC domain forms calcium-permeable channels in resistosomes; Activates downstream calcium signaling [17] |
Receptor-Like Kinases (RLKs)
RLKs are transmembrane proteins that contain an extracellular domain for ligand perception, a single transmembrane helix, and an intracellular kinase domain for signal transduction [13] [14]. They represent one of the largest gene families in plants, with over 600 members in Arabidopsis alone [18]. RLKs can be further categorized based on their extracellular domains, which include leucine-rich repeats (LRRs), lectin domains, and lysin motifs (LysMs) [3] [15].
The LRR-RLK subgroup is particularly important for plant immunity, with members such as FLS2 and EFR recognizing bacterial flagellin and elongation factor-Tu, respectively [15]. These receptors typically require co-receptors like BAK1 (BRASSINOSTEROID INSENSITIVE 1-ASSOCIATED RECEPTOR KINASE 1) for full functionality [12]. Upon ligand binding, RLKs undergo autophosphorylation and transphosphorylation events that activate downstream signaling cascades, including MAP kinase pathways and calcium-dependent signaling [13].
Table 2: Major RLK Subclasses in Plant Immunity
| RLK Subclass | Extracellular Domain | Recognized Ligands/PAMPs | Representative Examples | Function in Immunity |
|---|---|---|---|---|
| LRR-RLK-XII | Leucine-Rich Repeat (LRR) | Flagellin, EF-Tu, Xoo signals | FLS2, EFR, XA21 [15] | Pattern-triggered immunity against bacteria; Strong co-expansion with NLRs [15] |
| LysM-RLK | Lysin Motif (LysM) | Chitin, Peptidoglycan | CERK1 [15] | Fungal and bacterial cell wall component recognition; Mycorrhizal symbiosis [15] |
| LECRK | Lectin domain | Unknown carbohydrate motifs | LECRK family members [3] [11] | Cell death regulation; Damage-associated molecular pattern recognition [3] |
Receptor-Like Proteins (RLPs)
RLPs share structural similarities with RLKs, containing extracellular ligand-binding domains (often LRRs) and a transmembrane region, but lack a cytoplasmic kinase domain [18] [14]. In Arabidopsis, 57 LRR-RLPs have been identified and numbered consecutively according to their genomic positions [18]. RLPs require interaction with adaptor kinases such as SOBIR1 (SUPPRESSOR OF BIR1) and BAK1 to transduce intracellular signals [18].
Functionally, RLPs can be divided into two groups: those involved in development (e.g., RLP10/CLV2 and RLP17/TMM regulating meristem maintenance and stomatal patterning) and those participating in defense (e.g., RLP1, RLP23, RLP30, RLP32, and RLP42) [18]. Defense RLPs recognize a diverse array of pathogen-derived molecules, including NLP effectors, bacterial translation initiation factors, and fungal endopolygalacturonases [18]. Genomic analyses reveal that LRR-RLP gene families show a strong positive correlation with NLR expansion across plant species, suggesting functional coordination between these receptor types [15].
Computational Prediction of R-genes Using Deep Learning
Challenges in Traditional R-gene Identification
The identification and classification of R-genes present substantial challenges due to their unique genomic architecture, sequence diversity, and low sequence homology across species [3]. R-genes are frequently organized in clusters of closely duplicated genes within plant genomes, which complicates genome assembly and annotation processes [3]. Furthermore, their characteristically low expression levels make transcriptome-based prediction unreliable, and their similarity to repetitive sequences often leads to misannotation during standard genome annotation pipelines [3].
Traditional computational approaches for R-gene identification have primarily relied on alignment-based methods using tools such as BLAST, HMMER, and InterProScan to detect conserved protein domains characteristic of R-proteins [3] [14]. While these methods have been successful for identifying R-genes with high sequence similarity to known references, they frequently fail to detect divergent or novel R-genes with low homology to previously characterized sequences [3]. More recent machine learning approaches using support vector machines (SVMs) have improved prediction accuracy by extracting numerical features from protein sequences, but these still have limitations in capturing complex hierarchical patterns [3].
PRGminer: A Deep Learning Framework for R-gene Prediction
PRGminer represents a cutting-edge deep learning-based tool specifically designed for high-throughput prediction and classification of plant resistance genes [3] [11]. This tool implements a two-phase analytical framework that leverages the pattern recognition capabilities of deep neural networks to overcome limitations of traditional methods [3].
In Phase I, the system classifies input protein sequences as R-genes or non-R-genes using dipeptide composition features, achieving an impressive accuracy of 98.75% in k-fold validation and 95.72% on independent testing datasets with a Matthews correlation coefficient of 0.98 and 0.91, respectively [3]. Sequences classified as R-genes then proceed to Phase II, where they are categorized into one of eight distinct R-gene classes with an overall accuracy of 97.55% in k-fold testing and 97.21% on independent datasets [3] [11].
Table 3: PRGminer Classification System for R-gene Classes
| Class Code | Class Name | Key Domains/Features | Localization | Function |
|---|---|---|---|---|
| CNL | Coiled-coil-NBS-LRR | CC, NBS, LRR domains | Cytoplasmic | Effector-triggered immunity; Forms resistosomes [3] [11] |
| TNL | TIR-NBS-LRR | TIR, NBS, LRR domains | Cytoplasmic | ETI; TIR domain has NADase activity [3] [11] |
| RLK | Receptor-like kinase | eLRR, Kinase, TM domains | Plasma membrane | Pattern-triggered immunity; Signal transduction [3] [11] |
| RLP | Receptor-like protein | LRR, TM, short cytoplasmic tail | Plasma membrane | PAMP recognition; Requires adapter kinases [3] [11] |
| LYK | Lysin motif receptor kinase | LYSM, Kinase, TM domains | Plasma membrane | Chitin recognition; Fungal immunity [3] |
| LECRK | Lectin receptor-like kinase | Lectin, Kinase, TM domains | Plasma membrane | Carbohydrate binding; Cell death regulation [3] |
| KIN | Kinase | Kinase domain only | Cytoplasmic/ Membrane-associated | Signaling component; Phosphorylation cascades [3] |
| TIR | TIR domain | TIR domain only | Cytoplasmic | Signaling component; NADase activity [3] |
Experimental Protocols for R-gene Characterization
Protocol 1: Gain-of-Function Mutant Analysis for NLR Function
Purpose: To characterize the functional mechanisms of NLR-type R-genes using gain-of-function mutants that confer constitutive immune activation [16].
Background: The snc1 (suppressor of npr1-1, constitutive 1) mutant in Arabidopsis contains a single amino acid substitution (Glu552 to Lys) in the NL linker region of a TIR-NB-LRR protein, leading to constitutive defense activation without pathogen perception [16]. This system provides a valuable model for dissecting NLR signaling mechanisms.
Methods:
- Mutant Identification: Screen for constitutive defense response mutants displaying dwarf morphology, spontaneous lesion formation, and elevated PR gene expression in absence of pathogens [16].
- Genetic Mapping: Cross mutants with wild-type plants and utilize PCR-based molecular markers to map the mutation to specific chromosomal regions [16].
- Complementation Testing: Amplify candidate genes from mutant and wild-type plants and transform into mutant background to confirm genetic identity [16].
- Epistasis Analysis: Cross constitutive mutants with signaling component mutants (e.g., pad4-1, eds5-3) to determine genetic dependencies [16].
- Revertant Screening: Employ fast neutron bombardment to generate deletion mutants and identify revertants to confirm causal gene identity [16].
Key Applications: This approach enables researchers to identify critical regulatory domains within NLR proteins, elucidate signaling pathways downstream of NLR activation, and characterize the molecular switch mechanism controlling R-protein activity [16].
Protocol 2: Functional Characterization of RLPs Using Multi-omics
Purpose: To classify and characterize receptor-like proteins of unknown function as defense-related or developmental regulators using integrated multi-omics datasets [18].
Background: Among the 57 annotated RLPs in Arabidopsis, only a subset has known functions, with 6 validated in defense (VDRs) and 9 predicted as developmental orthologs (PDOs) based on comparative genomics [18]. This protocol enables systematic functional annotation of uncharacterized RLPs.
Methods:
- Transcriptome Analysis: Analyze publicly available RNA-seq datasets to compare expression patterns of target RLPs under pathogen infection, PAMP treatment, and across different tissues [18].
- Proteomic Profiling: Examine protein abundance data to identify correlations between transcript and protein levels, with defense RLPs typically showing lower abundance than developmental RLPs [18].
- Co-expression Network Analysis: Construct gene co-expression networks to identify potential interaction partners and functional modules [18].
- Genomic Distribution Assessment: Examine physical clustering of RLP genes in the genome, as defense-related RLPs often show tighter clustering [18].
- Diversity Analysis: Analyze pan-genome data to assess sequence diversity and selective pressures on different RLP subclasses [18].
Key Applications: This integrated approach facilitates the functional prediction of uncharacterized RLPs, identifies candidate immune receptors for genetic validation, and provides insights into the evolutionary dynamics of different RLP functional classes [18].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for R-gene Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Use |
|---|---|---|---|
| Mutant Lines | snc1, npr1-1, pad4-1, eds1, eds5-3 [16] | Genetic analysis of signaling pathways; Epistasis studies | Determine genetic dependencies and order of gene function in immune signaling [16] |
| Pathogen Strains | Pseudomonas syringae pv maculicola ES4326, Peronospora parasitica Noco2 [16] | Activation of specific R-gene pathways; Disease assays | Assess resistance specificity and strength of immune responses [16] |
| Signaling Mutants | bak1, sobir1 [18] | Disruption of receptor complex formation | Determine requirement for core signaling components in RLP function [18] |
| Domain Analysis Tools | InterProScan, HMMER, nCoil, Phobius, TMHMM2 [3] | Identification of protein domains and motifs | Structural annotation and classification of R-proteins [3] |
| Deep Learning Platforms | PRGminer webserver, Standalone package [3] [11] | High-throughput R-gene prediction and classification | Genome-wide identification and categorization of resistance genes [3] |
The structural and functional characterization of major R-gene classes has revealed remarkable diversity in plant pathogen recognition systems, yet also surprising coordination in their evolutionary trajectories. The finding that LRR-RLP and LRR-RLK-XII gene families show strong co-expansion with intracellular NLRs across 350 plant species suggests integrated evolution of the two-tiered plant immune system [15]. Deep learning tools like PRGminer represent a significant advancement in computational prediction of R-genes, achieving >95% accuracy in classification by leveraging dipeptide composition and domain features [3] [11].
Future research directions will likely focus on several key areas: (1) elucidating the structural mechanisms of resistosome formation and channel activity in NLR signaling; (2) understanding the molecular basis of integration between cell-surface and intracellular immune receptors; (3) leveraging deep learning approaches to predict R-gene function from sequence alone; and (4) harnessing this knowledge to engineer broad-spectrum, durable disease resistance in crop plants [17] [14] [15]. As genomic resources continue to expand, computational tools will play an increasingly vital role in accelerating the discovery and functional characterization of these essential components of plant immunity.
Plant resistance genes (R-genes) are fundamental components of the plant immune system, encoding proteins that detect pathogen invasion and initiate robust defense responses [3]. The identification of these genes is crucial for breeding disease-resistant crops and ensuring global food security. For decades, traditional genomics approaches have been the cornerstone of R-gene discovery. However, the complex genomic architecture of R-genes, particularly in economically important crops, presents significant challenges that limit the efficacy of these conventional methods [19] [20]. This application note examines the specific limitations of traditional genomics in R-gene identification and contextualizes these challenges within the broader research framework of PRGminer, a deep learning-based tool designed to overcome these obstacles [3] [11].
Core Limitations of Traditional Genomics in R-gene Identification
The application of traditional genomics to R-gene discovery encounters several fundamental obstacles that impact the completeness, accuracy, and efficiency of identification efforts.
Table 1: Key Limitations of Traditional Genomics in R-gene Identification
| Limitation Category | Specific Challenge | Impact on R-gene Discovery |
|---|---|---|
| Genomic Architecture | Gene clustering and sequence similarity [3] | Causes assembly issues and fragmented annotations |
| Technical Barriers | Low expression levels [3] | Difficulties in gene prediction from RNA-Seq data |
| Technical Barriers | Misidentification as repetitive elements [3] | Obscured R-gene loci during annotation |
| Methodological Constraints | Reliance on sequence homology [3] [14] | Failure to identify novel or divergent R-genes |
| Methodological Constraints | Limited domain recognition scope [14] | Incomplete classification of R-gene types |
| Data Complexity | Polyploid genomes [19] | Complications in gene annotation and analysis |
Structural and Technical Challenges
R-genes are frequently organized in clusters of closely related sequences within plant genomes. This arrangement creates substantial difficulties for local genome assembly processes, often resulting in incomplete and fragmented gene annotations [3]. The presence of numerous similar sequences can cause assembly algorithms to collapse these regions or produce incomplete representations. This challenge is particularly pronounced in polyploid species like sugarcane (Saccharum spp.) and Brassica napus, which contain multiple homologous genomes [19] [20]. In sugarcane, the combination of polyploidy and complex R-gene architecture necessitates specialized bioinformatics pipelines like DaapNLRSeek for accurate annotation [19].
Compounding these structural challenges, R-genes are typically expressed at low levels, making them difficult to detect and predict using standard RNA sequencing (RNA-Seq) approaches [3]. Furthermore, their repetitive nature often leads to misclassification as transposable elements during standard genome annotation processes that utilize public repeat databases, effectively causing these important defense genes to be masked in genomic analyses [3].
Methodological Constraints of Traditional Approaches
Traditional R-gene identification has heavily relied on alignment-based methods using tools such as BLAST, InterProScan, and HMMER3 to identify conserved domains and sequence similarities [3] [14]. While these approaches have successfully identified many known R-gene families, they possess an inherent limitation: their effectiveness diminishes substantially when targeting novel R-genes with low sequence homology to previously characterized genes [3]. This limitation is particularly problematic when studying wild plant species or crop wild relatives, which may contain valuable resistance genes with divergent sequences [3].
Table 2: Performance Comparison of R-gene Identification Methods
| Method Type | Examples | Key Advantages | Major Limitations |
|---|---|---|---|
| Alignment-Based | BLAST, HMMER, InterProScan [3] | Well-established, precise domain identification | Limited to known homologs; misses novel genes |
| Machine Learning | SVMProt-RF [21] | Better generalization than alignment-based | Limited feature extraction capability |
| Deep Learning | PRGminer [3] [11] | High accuracy (>98%); discovers novel patterns | Computational intensity; "black box" concerns |
Domain-based bioinformatics pipelines primarily focus on recognizing conserved structural motifs such as nucleotide-binding sites (NBS), leucine-rich repeats (LRRs), and coiled-coil (CC) domains [14]. While effective for classifying genes within known architectures, these methods may lack sensitivity in identifying atypical R-genes or those with unconventional domain combinations, potentially overlooking valuable resistance genes with novel structures [14].
Experimental Protocols for Traditional R-gene Identification
Protocol: Domain-Based R-gene Identification Using Sequence Homology
Application: Genome-wide identification of NBS-LRR resistance genes in plant genomes.
Reagents and Equipment:
- Genomic DNA or protein sequences
- High-performance computing cluster
- BLAST suite (v. 2.10+)
- HMMER software (v. 3.3+)
- InterProScan (v. 5.45+)
- Custom Perl/Python scripts for data parsing
Procedure:
- Sequence Database Preparation: Compile predicted protein sequences from the target genome assembly in FASTA format.
- Initial BLAST Screening: Perform BLASTp search against a curated database of known R-proteins using an E-value cutoff of 1e-5 [14].
- Domain Analysis: Process sequences through InterProScan to identify conserved R-gene domains (NBS, LRR, TIR, CC) [3].
- HMMER Scanning: Use hidden Markov model profiles from Pfam (NB-ARC: PF00931, TIR: PF01582, LRR: PF00560, CC: PF05725) for sensitive domain detection [14].
- Manual Curation: Visually inspect domain architecture and remove partial sequences or false positives.
- Classification: Categorize identified genes into CNL, TNL, RNL, RLK, RLP classes based on domain composition [14].
Technical Notes: This method typically achieves 70-80% accuracy for known R-gene families but struggles with divergent sequences and novel classes [21]. The process is time-intensive, requiring approximately 24-48 hours for a medium-sized plant genome (500 MB - 1 GB).
Protocol: Genome-Wide Association Study (GWAS) for R-gene Mapping
Application: Linking phenotypic resistance to genomic loci in plant populations.
Reagents and Equipment:
- Plant mapping population (F2, RILs, or natural diversity panel)
- DNA extraction kits
- Genotyping-by-sequencing or SNP array platform
- Phenotyping facilities with pathogen growth chambers
- TASSEL, GAPIT, or PLINK software
Procedure:
- Population Development: Create a segregating population of 200+ individuals from resistant × susceptible crosses.
- High-Density Genotyping: Generate SNP markers across the genome using appropriate genotyping platforms.
- Phenotypic Scoring: Inoculate plants with target pathogen and score disease symptoms using standardized scales.
- QTL Analysis: Perform interval mapping or association analysis to identify genomic regions associated with resistance.
- Fine Mapping: Develop additional markers in target regions to narrow candidate intervals.
- Candidate Gene Identification: Annotate genes in refined QTL regions and prioritize those with R-gene-like domains.
Technical Notes: This method is limited by population size, marker density, and heritability of the resistance trait. It typically identifies large genomic regions containing dozens to hundreds of genes, requiring substantial additional work for gene isolation [20].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for R-gene Identification Studies
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| PRGdb [14] | Curated database of known R-genes | Reference for alignment-based methods; contains experimentally validated genes |
| InterProScan [3] | Protein domain architecture analysis | Identifies conserved R-gene domains (NBS, LRR, TIR, CC) |
| HMMER3 [3] | Profile hidden Markov model search | Sensitive detection of divergent R-gene domains |
| Phobius [3] | Transmembrane topology prediction | Critical for identifying receptor-like proteins (RLPs) |
| SignalP [3] | Signal peptide prediction | Identifies secreted proteins and receptor extracellular domains |
| Pfam Database [3] | Collection of protein families | Provides HMM profiles for R-gene domains |
| NCBI RefSeq [3] | Reference sequence database | Source of annotated plant genomes for comparative analysis |
PRGminer: A Deep Learning Framework Overcoming Traditional Limitations
The PRGminer tool represents a paradigm shift in R-gene identification by employing a two-phase deep learning approach that circumvents many constraints of traditional methods [3] [11]. This tool achieves >98% accuracy in initial R-gene prediction and >97% accuracy in classifying R-genes into eight distinct structural classes, significantly outperforming alignment-based methods, particularly for novel or divergent sequences [3] [11].
Unlike traditional methods, PRGminer uses dipeptide composition and other sequence-derived features rather than relying on sequence homology, enabling it to identify R-genes based on underlying patterns rather than direct sequence similarity [3]. This approach eliminates problems associated with gene clustering, low expression, and repetitive element misidentification that plague traditional methods, as it operates directly on protein sequences without requiring complete genome assembly or expression data [3].
Traditional genomics approaches face substantial challenges in comprehensively identifying plant resistance genes due to the complex nature of R-gene architecture, limitations of homology-based methods, and technical barriers in genome assembly and annotation. While these methods continue to provide value for characterizing known R-gene families, their limitations in discovering novel resistance genes highlight the need for complementary approaches. Deep learning tools like PRGminer represent a promising alternative that transcends these limitations by leveraging pattern recognition capabilities that do not depend on sequence similarity. As plant pathogen pressures intensify due to climate change and agricultural intensification, overcoming these traditional limitations becomes increasingly critical for developing durable disease resistance in crop species.
The integration of deep learning (DL) into plant science is fundamentally transforming genomic prediction, enabling the accurate identification of complex genetic markers and resistance genes with unprecedented precision. This paradigm shift is particularly evident in plant disease resistance breeding, where tools like PRGminer leverage convolutional neural networks and multi-layer perceptrons to predict resistance genes (R-genes) far beyond the capabilities of traditional statistical methods. By processing high-dimensional genomic and phenomic data, DL models capture non-linear relationships and epistatic interactions that underlie complex traits such as disease resistance and yield. This document provides a comprehensive overview of key DL applications in plant genomics, detailed protocols for implementing tools such as PRGminer, and standardized workflows for genomic selection. These resources equip researchers with practical frameworks to harness DL technologies, accelerating the development of disease-resistant crops and enhancing global food security.
Plant diseases cause estimated annual yield losses of 20–30%, threatening global food security and necessitating the development of resistant crop varieties [14]. Traditional methods for identifying resistance genes (R-genes)—often reliant on alignment-based tools, manual phenotyping, and linear statistical models—are limited in scalability, throughput, and ability to model complex genetic architectures [3] [14]. The advent of deep learning (DL) has introduced a powerful alternative, capable of automatically learning hierarchical features from large-scale genomic, transcriptomic, and phenomic data.
DL architectures, including Convolutional Neural Networks (CNNs), Recurrent Neural Networks (RNNs), and Multi-Layer Perceptrons (MLPs), excel at capturing non-linear relationships and epistatic interactions among genomic markers [22] [23]. For example, MLPs have demonstrated superior performance in predicting disease resistance and yield traits in wheat, maize, and rice compared to the traditional Genomic Best Linear Unbiased Predictor (GBLUP) [22]. The development of specialized tools such as PRGminer exemplifies this shift, employing deep learning to accurately classify R-genes and distinguish among various R-gene families based on protein sequences [3] [24].
This article outlines major application areas of DL in plant genomic prediction, provides a detailed experimental protocol for the PRGminer tool, and presents a generalized DL workflow for genomic selection. Target audiences include plant biologists, bioinformaticians, and breeders seeking to implement DL frameworks for crop improvement.
Key Applications of Deep Learning in Genomic Prediction
Deep learning applications in plant science extend across genomics, phenomics, and multi-omics data integration. The table below summarizes major DL architectures and their target applications in plant genomic prediction.
Table 1: Key Deep Learning Architectures and Applications in Plant Genomic Prediction
| Deep Learning Architecture | Primary Application Area | Key Functionality | Reported Performance/Impact |
|---|---|---|---|
| Multi-Layer Perceptron (MLP) [22] | Genomic Selection (GS) | Predicts complex agronomic traits from genome-wide markers; captures non-linear and epistatic interactions. | Frequently outperforms GBLUP, especially for complex traits in smaller datasets (n < 1,000). [22] |
| Convolutional Neural Network (CNN) [3] [25] | R-gene Identification & Classification | Processes protein sequences and image-based phenomic data; extracts hierarchical features for classification. | PRGminer (using dipeptide composition) achieved 98.75% k-fold accuracy in R-gene identification. [3] [24] |
| Multi-Task Learning (MTL) Models [26] | Multi-Trait Genomic Prediction | Simultaneously learns multiple correlated phenotypes using shared parameters and task-specific networks. | MtCro model showed 1-9% performance gain over single-task models on wheat and maize datasets. [26] |
| Support Vector Machine (SVM) [27] | Resistance Gene Identification | Classifies resistant/susceptible varieties based on transcriptomic expression profiles of key genes. | An SVM model trained on 24 key genes achieved a classification accuracy of 0.9514 for sunflower broomrape resistance. [27] |
| Large Multimodal Models (LMMs) [25] | Integrated Disease Diagnosis | Interprets complex disease patterns by fusing heterogeneous data (e.g., imagery, genomics, text). | Emerging technology with groundbreaking potential for holistic phenotype prediction and interpretation. [25] |
These architectures are being deployed to overcome specific challenges in plant breeding. For instance, MLPs address the limitations of linear models like GBLUP in capturing the complex genetic architecture of disease resistance, often demonstrating superior predictive accuracy [22]. CNNs form the backbone of specialized R-gene discovery tools like PRGminer, which automates the identification and classification of resistance proteins from sequence data with high accuracy [3]. Beyond single-task prediction, multi-task learning frameworks such as MtCro leverage genetic correlations between different phenotypes (e.g., yield and thousand kernel weight) to improve prediction accuracy for all traits simultaneously and enhance breeding efficiency [26].
Application Note: The PRGminer Tool for R-gene Prediction
PRGminer is a deep learning-based tool specifically designed for the high-throughput prediction and classification of plant resistance genes (R-genes) from protein sequences [3]. It addresses the challenges of identifying R-genes in newly sequenced or wild plant species, where traditional similarity-based methods often fail due to low sequence homology [3]. The tool is implemented as a two-phase classification system and is available via a freely accessible webserver or as a standalone tool from a GitHub repository [3] [9].
Operational Protocol
The following protocol outlines the standard workflow for using the PRGminer webserver.
Table 2: PRGminer Research Reagent Solutions
| Item Name | Specification / Type | Critical Function in the Workflow |
|---|---|---|
| Input Protein Sequence(s) | FASTA format | Serves as the primary data for R-gene prediction and classification. |
| Dipeptide Composition Encoding | Feature extraction method | Converts protein sequences into a numerical representation optimal for the deep learning model. [3] |
| Convolutional Neural Network (CNN) | Deep learning architecture | Automatically extracts relevant features from the encoded sequences for accurate classification. |
| Web Server / Standalone Package | Platform | Provides the user interface and computational backend for performing predictions. [9] |
Procedure:
- Input Preparation: Provide the protein sequence(s) for analysis using one of three accepted methods:
- Accession ID: Enter a valid NCBI or UniProt protein accession ID.
- FASTA File Upload: Upload a text file containing one or multiple protein sequences in FASTA format.
- Direct Pasting: Paste the FASTA-formatted sequence(s) directly into the provided text area [9].
- Job Submission: Initiate the analysis by clicking the "Run Prediction" button. The system will process the input sequences through its two-phase DL pipeline [9].
- Results Interpretation: After processing, the results are displayed in a table and available for download. Key outputs include:
- Phase I Result: A binary prediction indicating whether the protein is an "R-gene" or "Non-R-gene."
- Confidence Score: A probabilistic score reflecting the model's confidence in the Phase I prediction.
- Phase II Result (If applicable): For sequences predicted as R-genes, a classification into one of eight specific classes (e.g., CNL, TNL, RLK, RLP) is provided [3] [9].
The following diagram illustrates the logical workflow and two-phase architecture of the PRGminer tool.
Performance and Validation
In independent testing, PRGminer demonstrated an accuracy of 95.72% in Phase I (R-gene identification) and 97.21% in Phase II (R-gene classification), with Matthews Correlation Coefficient (MCC) values of 0.91 and 0.92, respectively, indicating robust performance beyond random chance [3] [24]. The tool has been rigorously validated on experimentally confirmed R-genes, confirming its efficacy in accelerating the discovery of novel resistance genes for breeding programs [3].
General Protocol: A DL Workflow for Genomic Selection
This protocol describes a standardized workflow for implementing a deep learning-based genomic selection (GS) pipeline, adaptable for predicting traits like disease resistance or yield.
Procedure:
Dataset Curation:
- Genotypic Data: Obtain genome-wide marker data (e.g., SNPs) for a population of plant lines. Code the genotypes numerically (e.g., 0 for AA, 1 for AT, 2 for TT) [26].
- Phenotypic Data: Collect corresponding phenotypic measurements (e.g., disease severity scores, yield) for the same lines. For multi-trait prediction, compile data for all target traits [26].
- Data Preprocessing: Perform quality control on markers (e.g., remove low-frequency SNPs), impute missing genotypes, and consider using Principal Component Analysis (PCA) for dimensionality reduction if required by the model [26].
Model Selection and Configuration:
- Architecture Choice: Select an appropriate DL architecture. A Multi-Layer Perceptron (MLP) is a strong starting point for genomic prediction [22].
- Model Design: For multi-trait prediction, implement a multi-task learning framework like MtCro, which uses a shared-bottom network to learn from all traits simultaneously and task-specific tower networks for final trait-specific predictions [26].
- Hyperparameter Tuning: Meticulously tune hyperparameters (e.g., number of layers and units, learning rate, dropout rate) specific to the dataset to maximize predictive accuracy [22].
Model Training and Validation:
- Data Splitting: Partition the dataset into training, validation, and independent testing sets.
- Training: Train the model on the training set, using the validation set for early stopping to prevent overfitting.
- Performance Assessment: Evaluate the final model on the held-out test set using metrics such as Pearson's correlation coefficient between predicted and observed values, mean squared error, and prediction accuracy [22] [26].
The following diagram summarizes this generalized genomic selection workflow.
Deep learning is undeniably reshaping the landscape of genomic prediction in plant science. By moving beyond the constraints of linear models, DL enables the accurate prediction of complex traits and the high-throughput discovery of crucial resistance genes, as powerfully demonstrated by the PRGminer tool. The provided application notes and standardized protocols offer a practical foundation for researchers to integrate these advanced computational methods into their breeding programs. As these technologies continue to evolve, their integration with multi-omics data and field-based phenomics will be critical for developing next-generation crops with durable disease resistance and enhanced climate resilience, thereby securing future global food supplies.
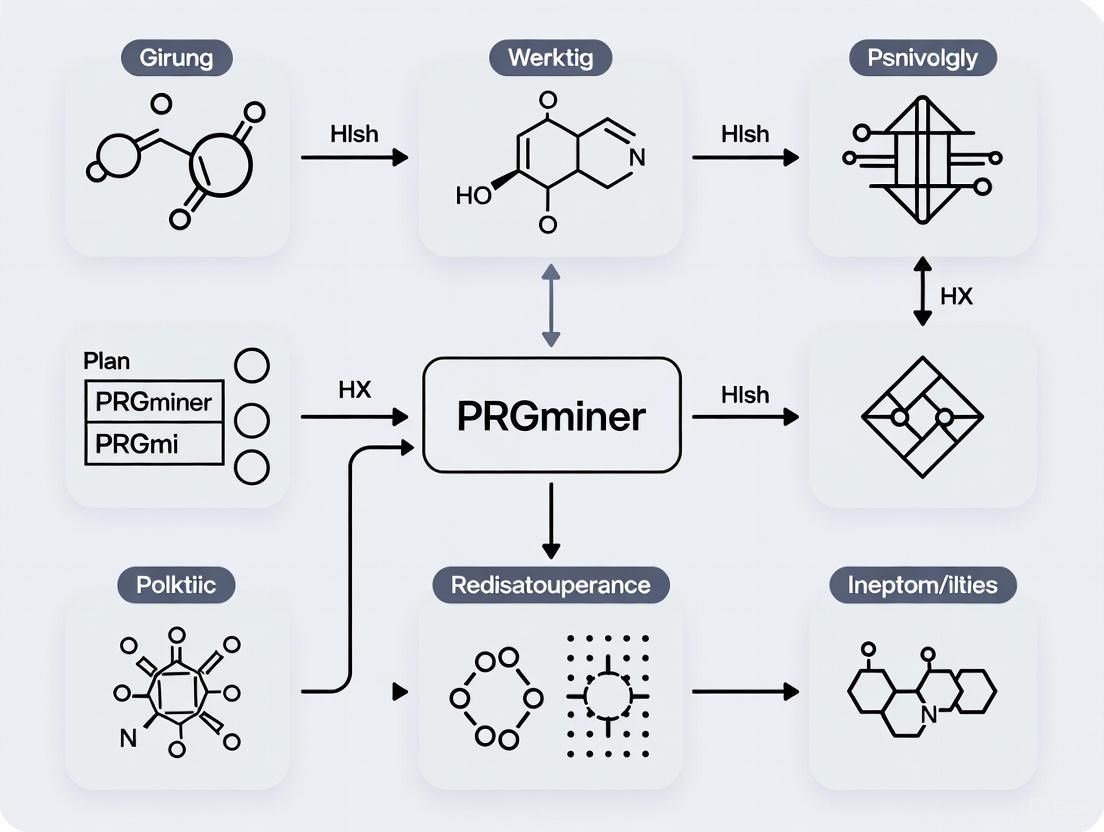
A Deep Dive into PRGminer: Architecture and Practical Workflow
In the broader context of deep learning prediction of plant resistance genes, the PRGminer tool represents a significant advancement in computational biology for agricultural science. Accurately identifying plant resistance (R) genes is a critical component of disease resistance breeding, as these genes encode proteins that identify specific molecular patterns associated with invading pathogens [3]. When activated, R-genes initiate molecular processes that activate defensive responses including synthesis of antimicrobial compounds, cell wall strengthening, and programmed cell death in infected cells [3]. Traditional methods for identifying R-genes in wild species and near relatives of plants are both challenging and time-consuming, creating bottlenecks in crop improvement programs [3]. PRGminer addresses these limitations through a specialized two-phase deep learning framework that enables high-throughput prediction and classification of resistance genes from protein sequences, offering researchers a powerful tool to accelerate the discovery of new R-genes and develop strategies for breeding disease-resistant plants [3].
The Computational Architecture of PRGminer
PRGminer implements a sophisticated two-phase analytical framework for comprehensive resistance gene characterization. Phase I performs binary classification, predicting whether input protein sequences are R-genes or non-R-genes [3]. This initial filtering step ensures that only genuine resistance genes proceed to further analysis. Sequences classified as non-R-genes are excluded from subsequent processing, while those identified as R-genes advance to Phase II, where they undergo detailed categorization into specific resistance gene classes based on their domain architectures and functional characteristics [3] [11].
This sequential approach mirrors the logical workflow that plant pathologists and breeders would follow when characterizing resistance genes—first identifying candidate sequences from genomic data, then determining their specific functional classifications. The implementation of this process through deep learning rather than traditional alignment-based methods allows PRGminer to identify resistance genes even in cases of low sequence homology, which is particularly valuable when annotating newly sequenced plant genomes [3].
Workflow Visualization
The following diagram illustrates PRGminer's integrated two-phase prediction workflow:
Figure 1: PRGminer's two-phase prediction workflow. Phase I filters protein sequences, identifying R-genes versus non-R-genes. Phase II classifies confirmed R-genes into eight specific classes.
Phase I: R-gene Identification
Experimental Protocol & Methodology
The Phase I prediction module utilizes a deep learning framework trained on comprehensive protein sequence datasets. The training data for PRGminer development was obtained from various public databases including Phytozome, Ensemble Plants, and NCBI to ensure broad coverage of known resistance genes and non-R-gene sequences [3]. During development, researchers tested multiple sequence representations and found that dipeptide composition provided the optimal predictive performance [3].
The implementation employs deep learning algorithms that extract both sequential and convolutional features from raw encoded protein sequences, moving beyond traditional alignment-based methods that often fail with sequences having low homology [3]. This approach allows the model to identify patterns and features indicative of resistance genes that might be missed by conventional similarity-based methods such as BLAST, InterProScan, or HMMER [3].
For users, the Phase I analysis requires submitting protein sequences through one of three input methods: (1) entering a valid protein accession ID from NCBI or UniProt, (2) uploading a FASTA file containing single or multiple protein sequences, or (3) directly pasting FASTA-formatted sequences into the text area [9]. The system then processes these sequences through its trained model to generate prediction outcomes.
Performance Metrics
Table 1: Performance metrics of PRGminer's Phase I R-gene prediction
| Metric | k-fold Training/Testing | Independent Testing |
|---|---|---|
| Accuracy | 98.75% | 95.72% |
| Matthews Correlation Coefficient | 0.98 | 0.91 |
The performance metrics demonstrate that Phase I achieves exceptionally high prediction accuracy, with a Matthews correlation coefficient of 0.98 during k-fold validation, indicating robust model performance [3]. The maintained high accuracy (95.72%) on independent testing datasets confirms the model's generalizability beyond its training data [3].
Phase II: R-gene Classification
Experimental Protocol & Methodology
Phase II of PRGminer implements a multi-class classification system that categorizes resistance genes confirmed in Phase I into eight specific classes based on their domain architectures and functional characteristics. This classification is essential for understanding the potential mechanisms of resistance and guiding further functional characterization.
The deep learning model in Phase II was trained to recognize the distinctive protein domains and structural features that define each R-gene class. The system analyzes the sequence characteristics and patterns that correspond to specific domain combinations, allowing accurate classification without requiring explicit domain annotation for each sequence.
Users automatically advance sequences classified as R-genes in Phase I to Phase II analysis, where they receive detailed classification results indicating the specific R-gene category along with confidence metrics for each prediction.
Resistance Gene Classes
Table 2: Classification schema for plant resistance genes in PRGminer Phase II
| Class | Domain Architecture | Functional Role |
|---|---|---|
| CNL | Coiled-coil, Nucleotide-binding site, Leucine-rich repeat | Cytosolic resistance receptor; recognizes pathogen effectors and triggers immune response [11] |
| TNL | Toll/interleukin-1 receptor, NBS, LRR | Cytosolic resistance receptor; contains TIR domain at N-terminus instead of coiled-coil [11] |
| RLP | Leucine-rich repeat, Transmembrane domain, Short cytoplasmic region | Membrane-bound receptor; recognizes avirulence genes indirectly, often through partner proteins [11] |
| RLK | Extracellular leucine-rich repeat, Kinase domain | Membrane-bound receptor; eLRR domain recognizes ligands, kinase domain triggers downstream signaling [11] |
| LECRK | Lectin, Kinase, Transmembrane domains | Lectin receptor-like kinase involved in resistance processes [11] |
| LYK | Lysin motif, Kinase, Transmembrane domains | Lysin motif receptor kinase; recognizes specific molecular patterns [11] |
| TIR | Toll/interleukin-1 receptor domain | Contains TIR domain only, lacking LRR or NBS domains [11] |
| KIN | Kinase domain | Kinase domain involved in resistance process [11] |
Performance Metrics
Table 3: Performance metrics of PRGminer's Phase II R-gene classification
| Metric | k-fold Training/Testing | Independent Testing |
|---|---|---|
| Overall Accuracy | 97.55% | 97.21% |
| Matthews Correlation Coefficient | 0.93 | 0.92 |
Phase II maintains exceptional classification accuracy exceeding 97% on both k-fold validation and independent testing, with consistently high Matthews correlation coefficients around 0.92-0.93 [3]. This performance demonstrates the model's reliability in distinguishing between the eight resistance gene classes with high precision.
The Researcher's Toolkit for PRGminer Implementation
Table 4: Essential research reagents and computational resources for PRGminer implementation
| Resource | Type | Function/Purpose | Access Information |
|---|---|---|---|
| PRGminer Web Server | Web Application | Primary interface for sequence prediction and classification | Freely accessible at: https://kaabil.net/prgminer/ [3] |
| Standalone PRGminer Tool | Software Package | Local installation for large-scale analyses or pipeline integration | Download from: https://github.com/usubioinfo/PRGminer [3] |
| NCBI Protein Database | Data Resource | Source of protein sequences for analysis and validation | https://www.ncbi.nlm.nih.gov/ [3] |
| UniProt | Data Resource | Alternative source of protein sequences with comprehensive annotation | https://www.uniprot.org/ [9] |
| Phytozome | Data Resource | Plant genomic data for context and validation | https://phytozome-next.jgi.doe.gov/ [3] |
| Ensemble Plants | Data Resource | Plant genomic data for context and validation | https://plants.ensembl.org/ [3] |
Implementation Guide
For researchers implementing PRGminer in their resistance gene discovery workflows, several practical considerations ensure optimal results. The web server is recommended for most individual users and small-scale analyses, with typical processing times of approximately two minutes for standard datasets [11]. The standalone tool is preferred for large-scale analyses involving more than 10,000 sequences, integration with existing bioinformatics pipelines, customized analytical workflows, or offline usage scenarios [9].
Local installation requires Python 3.7 or higher and specific dependencies detailed in the requirements.txt file included with the distribution [9]. While GPU support is optional, it significantly accelerates processing for large datasets. The input requirements are flexible, accepting protein sequences in standard FASTA format through multiple submission methods [9].
Technical Specifications and System Capabilities
Performance and Scalability
PRGminer demonstrates exceptional computational efficiency and scalability characteristics that make it suitable for both individual research projects and large-scale genomic screening initiatives. The system achieves high efficiency in processing large protein sequence datasets through optimized computational pipelines [11]. This efficiency is maintained across diverse dataset sizes, from individual sequences to genome-scale analyses.
The tool's architecture is designed for high scalability, enabling researchers to process extensive genomic datasets without performance degradation [11]. This capability is particularly valuable for comprehensive genome-wide identification of resistance genes across major crops, which has become increasingly important in crop improvement programs [14].
Integration with Research Workflows
PRGminer functions effectively as both a standalone prediction tool and as a component within broader bioinformatics pipelines for plant resistance gene discovery. The system complements existing computational approaches for identifying resistance proteins, including traditional domain-based methods, machine learning classifiers, and specialized R-gene databases [14]. The integration of PRGminer's deep learning capabilities with established resources such as PRGdb, PlantNLRatlas, and RefPlantNLR creates powerful synergies that accelerate the identification of novel R-proteins and deepen our understanding of plant immunity [14].
The following diagram illustrates the classification system for plant resistance genes, showing the relationships between major categories:
Figure 2: Classification hierarchy of plant resistance genes. PRGminer categorizes R-genes into intracellular receptors, membrane-bound receptors, and other classes based on domain architecture.
PRGminer's two-phase prediction engine represents a significant advancement in computational methods for plant resistance gene discovery. By integrating deep learning with specialized biological domain knowledge, the tool provides researchers with an accurate and efficient system for both identifying and classifying resistance genes from protein sequences. The exceptional performance metrics demonstrated across both phases of analysis—with accuracies exceeding 95% in independent testing—validate the robustness of this approach for accelerating plant resistance gene research [3].
The tool's accessibility through both web server and standalone application ensures broad usability across the research community, from individual investigators to large-scale crop improvement programs. As the field of plant pathology increasingly embraces computational methods for understanding disease resistance mechanisms [14], PRGminer offers a sophisticated solution that bridges the gap between traditional bioinformatics and modern deep learning approaches. This integration is particularly valuable for addressing the persistent challenge of developing disease-resistant crops in the face of evolving pathogen threats, ultimately contributing to more sustainable agricultural practices and enhanced global food security.
The identification of plant resistance (R) genes is a critical component in the effort to safeguard global food security. These genes encode proteins that enable plants to detect specific pathogen-derived molecular patterns and initiate robust immune responses [3]. Conventional methods for R-gene identification, which often rely on sequence alignment and domain homology, are challenged by the immense diversity and rapid evolution of these genes, making the process time-consuming and often inadequate for characterizing novel resistance traits [3] [14].
Deep learning models present a powerful alternative by learning complex sequence-function relationships directly from primary protein data. A key to harnessing this power lies in the effective numerical representation of protein sequences. This application note elucidates the central role of dipeptide composition as a feature encoding strategy within the PRGminer deep learning tool, providing a detailed protocol for its implementation in the prediction and classification of plant resistance genes [3].
The PRGminer Framework: A Two-Phase Prediction System
PRGminer is engineered as a high-throughput, deep learning-based tool that operates through two sequential analytical phases to ensure precise identification and functional categorization of resistance genes [3].
- Phase I: R-gene vs. Non-R-gene Identification: This initial classification layer acts as a filter, distinguishing potential
R-genes from all other input protein sequences. A sequence classified as a "non-R-gene" is excluded from further analysis. - Phase II: R-gene Functional Classification: Protein sequences identified as
R-genes in Phase I are subsequently classified into one of eight major categories based on their specific domain architectures and presumed functional mechanisms [3].
The workflow is designed for efficiency and accuracy, ensuring that only high-confidence R-gene candidates undergo detailed subtyping. Figure 1 illustrates this sequential analytical process.
Figure 1. The two-phase workflow of PRGminer for R-gene identification and classification.
Core Methodology: Feature Encoding with Dipeptide Composition
The performance of a deep learning model is heavily dependent on how biological sequences are converted into numerical features. PRGminer leverages dipeptide composition (DipC) as a primary feature encoding method, which was found to yield superior prediction performance compared to other representations [3].
Protocol: Calculating Dipeptide Composition
This protocol details the steps to convert a raw protein sequence into a fixed-length dipeptide composition feature vector.
Principle: Dipeptide composition encapsulates the fractional frequencies of all 400 possible adjacent amino acid pairs (e.g., Ala-Ala, Ala-Cys, Ala-Asp... Trp-Trp) within a protein sequence. This provides a global representation of local amino acid arrangement patterns, capturing critical information about residue correlations that is lost in single amino acid composition [3] [28].
Materials:
- Input Data: Protein sequence(s) in FASTA format.
- Computing Environment: A Python environment (v3.7 or higher) with the
NumPylibrary.
Procedure:
- Sequence Preprocessing: Obtain the canonical protein sequence. Remove any non-standard amino acid characters or ambiguous residues to ensure the sequence is composed only of the 20 standard amino acids.
- Generate All Possible Dipeptides: Define the list of all 400 possible dipeptides from the 20 standard amino acids.
- Count Dipeptide Occurrences: Traverse the input protein sequence from the N- to C-terminus, counting the occurrence of every dipeptide. For a sequence of length
L, there will beL-1dipeptides.- Example: For a sequence "MAGK", the dipeptides are "MA", "AG", and "GK".
- Calculate Fractional Frequencies: Normalize the count of each dipeptide by the total number of dipeptides in the sequence (
L-1). This generates the composition vector, which is independent of sequence length.- Formula:
Frequency(Dipeptide_i) = (Count(Dipeptide_i) / (L-1)) * 100
- Formula:
- Vector Construction: Construct a fixed-length feature vector of 400 dimensions, where each dimension corresponds to the normalized frequency of one of the 400 dipeptides.
This process results in a normalized, length-independent numerical representation of the protein sequence that is suitable for input into a deep learning model. Table 1 provides a comparative overview of the performance achieved by different feature encoding methods within the PRGminer framework.
Table 1: Performance comparison of feature encoding methods in PRGminer's Phase I (R-gene vs. Non-R-gene) prediction. Metrics are based on independent testing as reported in the original study [3].
| Feature Encoding Method | Prediction Accuracy (%) | Matthews Correlation Coefficient (MCC) |
|---|---|---|
| Dipeptide Composition (DipC) | 95.72 | 0.91 |
| Other Representational Methods | Lower | Lower |
Model Architecture and Performance
PRGminer utilizes a deep learning architecture, specifically a Convolutional Neural Network (CNN), to learn from the dipeptide-encoded protein sequences. CNNs are adept at identifying local, informative patterns within spatial data—in this case, the patterns of dipeptide frequencies that are characteristic of resistance genes and their subclasses [3] [14].
The model processes the 400-dimensional dipeptide vector through multiple layers to automatically learn hierarchical features. Lower layers may detect simple motifs, while higher layers combine these into more complex functional representations relevant to pathogen recognition and immune signaling. Figure 2 provides a simplified schematic of this deep learning architecture.
Figure 2. Schematic of the deep learning model in PRGminer that processes dipeptide composition inputs.
The integration of dipeptide composition with this deep learning model has yielded state-of-the-art prediction performance. Table 2 summarizes the final performance metrics of PRGminer across its two operational phases, demonstrating high accuracy and reliability.
Table 2: Overall performance metrics of PRGminer's two-phase prediction system. Data is sourced from the original research, which employed k-fold cross-validation and independent testing [3].
| Phase | Description | k-fold Testing Accuracy (%) | Independent Testing Accuracy (%) | Independent Testing MCC |
|---|---|---|---|---|
| I | R-gene vs. Non-R-gene | 98.75 | 95.72 | 0.91 |
| II | R-gene Classification | 97.55 | 97.21 | 0.92 |
The Scientist's Toolkit: Research Reagent Solutions
The following table details key computational tools and resources that are essential for research in the field of deep learning-based R-gene prediction, including those utilized by PRGminer and comparable approaches.
Table 3: Essential research reagents and computational tools for R-gene prediction.
| Item Name | Type/Function | Relevance in R-gene Research |
|---|---|---|
| PRGminer Web Server | Accessible Prediction Tool | Provides a user-friendly interface for submitting protein sequences and receiving R-gene predictions and classifications, requiring no local installation [3]. |
| PRGminer Standalone Code | Downloadable Software | Allows researchers to run and potentially customize the PRGminer tool on their own computational infrastructure for large-scale or proprietary analyses [3]. |
| InterProScan | Domain & Motif Detection | A foundational, alignment-based tool for identifying functional domains and motifs in protein sequences; used by many traditional R-gene prediction pipelines for comparative analysis [14]. |
| Phytozome/Ensemble Plants | Genomic Database | Repositories of plant genomic data that serve as primary sources for obtaining protein sequences used in training and testing deep learning models like PRGminer [3]. |
| LASSO Regression | Feature Selection Algorithm | A machine learning method used to identify the most informative biomarkers (e.g., key genes) from high-dimensional data, as demonstrated in other plant resistance studies [27]. |
PRGminer is a deep learning-based high-throughput tool specifically designed for the identification and classification of plant resistance genes (R-genes) [3]. Accurately identifying R-genes in wild species and near relatives of plants is challenging and time-consuming using conventional methods [3]. PRGminer addresses this challenge through a robust two-phase prediction system, achieving high accuracy in both phases [3] [24]. This guide provides detailed protocols for utilizing both the web server and standalone tool, enabling researchers to efficiently integrate PRGminer into their plant resistance gene discovery pipelines.
PRGminer Web Server Protocol
The PRGminer web server provides a user-friendly interface for researchers to analyze protein sequences without requiring local installation or computational expertise.
Input Submission Methods
The web server accepts protein sequences through three primary input methods [9]:
- Accession ID: Enter a valid protein accession ID from NCBI or UniProt to automatically fetch and analyze the sequence.
- FASTA File Upload: Upload a FASTA file containing one or multiple protein sequences for batch analysis.
- Direct Sequence Pasting: Paste FASTA-formatted sequences directly into the provided text area.
Execution and Output Interpretation
After selecting your preferred input method, click "Run Prediction" to initiate the analysis [9]. The typical processing time is approximately two minutes [11]. Results are presented in a structured table format containing [9]:
- Sequence ID and basic information
- Prediction outcome (R-gene or Non-R-gene)
- Confidence scores for predictions
- Detailed classification for R-genes
The results table provides downloadable options in CSV, JSON, or FASTA formats, including sequences and predictions. Users can also download filtered results for specific R-gene classes or confidence thresholds [9].
Standalone Tool Installation and Usage
For large-scale analyses or integration into existing bioinformatics pipelines, the standalone version of PRGminer is recommended.
System Requirements and Installation
The standalone tool requires specific system configuration for optimal performance [9]:
Table: System Requirements for Standalone PRGminer
| Component | Minimum Requirement | Recommended Specification |
|---|---|---|
| Python Version | Python 3.7 or higher | Python 3.8+ |
| Memory | Sufficient RAM for datasets | High RAM for large datasets (>10,000 sequences) |
| Processing | Standard CPU | GPU support (for faster processing) |
| Usage | Basic command line | Integration with existing pipelines |
Local installation is particularly recommended for processing large datasets exceeding 10,000 sequences, integration with existing pipelines, customized analysis workflows, and offline usage [9]. The standalone tool is available for download at https://github.com/usubioinfo/PRGminer [3].
Command Line Operations
After installation, users can execute PRGminer via command line interface. The tool maintains the same two-phase analysis approach as the web server, processing input sequences through initial R-gene identification followed by detailed classification.
Experimental Design and Performance Metrics
PRGminer implements a sophisticated two-phase deep learning framework for R-gene prediction and classification.
Two-Phase Analysis Workflow
The analytical process consists of two distinct phases [3]:
- Phase I - R-gene Prediction: Classifies input protein sequences as R-genes or non-R-genes using dipeptide composition features.
- Phase II - R-gene Classification: Further classifies predicted R-genes into eight specific classes based on domain architecture.
Performance Validation
PRGminer has demonstrated exceptional performance during validation studies, achieving high accuracy in both phases of analysis [3] [24]:
Table: PRGminer Performance Metrics
| Phase | Evaluation Method | Accuracy | MCC Value |
|---|---|---|---|
| Phase I | k-fold training/testing | 98.75% | 0.98 |
| Phase I | Independent testing | 95.72% | 0.91 |
| Phase II | k-fold training/testing | 97.55% | 0.93 |
| Phase II | Independent testing | 97.21% | 0.92 |
The high Matthews Correlation Coefficient (MCC) values indicate robust predictive performance across both balanced and imbalanced datasets [3].
R-gene Classification System
PRGminer classifies resistance genes into eight distinct categories based on their domain architectures and functional characteristics [11].
Domain Architectures and Functions
The classification system encompasses major R-gene classes with their specific domain compositions:
Biological Significance of R-gene Classes
Each R-gene class plays distinct roles in plant immunity, recognizing different pathogen-associated molecular patterns and initiating specific defense responses [3] [11]. Understanding these classifications helps researchers interpret PRGminer results in the context of plant defense mechanisms.
Research Reagent Solutions
Successful implementation of PRGminer requires specific computational resources and biological data sources.
Table: Essential Research Reagents and Resources
| Reagent/Resource | Function/Purpose | Source/Example |
|---|---|---|
| Protein Sequences | Input data for R-gene prediction | Phytozome, Ensemble Plants, NCBI [3] |
| FASTA Formatted Files | Standard format for sequence submission | Custom datasets, public repositories |
| Deep Learning Framework | Backend for prediction algorithms | TensorFlow/PyTorch implementation [24] |
| Computational Resources | Hardware for standalone tool execution | CPU/GPU systems with sufficient RAM [9] |
| Validation Datasets | Performance assessment and benchmarking | Experimentally validated R-gene datasets [3] |
Application in Plant Resistance Research
PRGminer significantly accelerates the discovery of novel resistance genes, enabling researchers to understand the genetic basis of plant immunity and develop strategies for breeding disease-resistant crops [3]. The tool's high accuracy in identifying diverse R-gene classes makes it particularly valuable for studying resistance mechanisms in newly sequenced plant genomes where traditional similarity-based methods often fail due to low homology [3]. By integrating PRGminer into their research pipelines, scientists can efficiently screen large genomic datasets, identify potential resistance candidates, and prioritize targets for experimental validation.
PRGminer is a deep learning-based tool designed for the high-throughput prediction and classification of plant resistance genes (R-genes). Its analytical process is implemented in two distinct, sequential phases to ensure accurate and detailed results for researchers [3] [8]. The tool addresses a critical need in plant science, as the identification of new R-genes in wild species and relatives is both challenging and time-consuming, yet essential for disease resistance breeding [3]. Proper interpretation of its outputs—specifically the confidence scores and the subsequent eight-class categorization—is fundamental to leveraging its full potential in understanding plant defense mechanisms and guiding breeding strategies.
The following workflow diagram illustrates the two-phase prediction process of PRGminer, from input to final classification:
Phase I: R-gene vs. Non-R-gene Prediction
Performance Metrics and Confidence Score Interpretation
The first phase of PRGminer acts as a binary classifier, determining whether a query protein sequence is a resistance gene (R-gene) or a non-resistance gene (non-R-gene) [3] [8]. The model's performance on an independent testing set provides the basis for trusting its predictions, as summarized in the table below.
Table 1: Performance Metrics of PRGminer in Phase I (Binary Classification)
| Metric | k-fold Training/Testing Performance | Independent Testing Performance |
|---|---|---|
| Accuracy | 98.75% | 95.72% |
| Matthews Correlation Coefficient (MCC) | 0.98 | 0.91 |
The confidence score generated in Phase I is a probabilistic value between 0 and 1, reflecting the model's certainty that the input sequence is an R-gene. A score closer to 1 indicates high confidence in an R-gene prediction, while a score closer to 0 indicates high confidence in a non-R-gene classification. The high MCC value of 0.91 on the independent test set is particularly noteworthy. The MCC is considered a robust metric for binary classifications, especially on imbalanced datasets, as it accounts for true and false positives and negatives [3] [8]. An MCC value this close to 1 signifies an almost perfect prediction model, giving researchers high confidence in the tool's binary output.
Experimental Protocol for Phase I Validation
Objective: To validate the binary classification performance of PRGminer's Phase I model. Input: A set of protein sequences (e.g., 10% of the total dataset, withheld from training). Method:
- Input Preparation: Format query protein sequences in FASTA format.
- Model Loading: Load the pre-trained Phase I deep learning model, which uses dipeptide composition for sequence representation.
- Prediction Execution: Submit the sequences to the PRGminer webserver or standalone tool.
- Output Collection: Record the binary prediction (R-gene/Non-R-gene) and the associated confidence score for each sequence.
- Performance Calculation: Compare predictions against known labels to calculate:
- Accuracy: (True Positives + True Negatives) / Total Predictions
- MCC: Calculated from the confusion matrix, providing a balanced measure.
Phase II: Eight-Class R-gene Categorization
Classification System and Performance
Sequences identified as R-genes in Phase I proceed to Phase II, where they are classified into one of eight specific categories based on their protein domain architecture [3]. This detailed classification is crucial because different R-gene classes are involved in distinct layers of the plant immune system, such as effector-triggered immunity (ETI) and pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) [14] [29].
The following diagram illustrates the hierarchical relationship between the major plant immunity layers and the eight R-gene classes predicted by PRGminer:
The performance of this multi-class classification is robust, as shown in the table below.
Table 2: Performance Metrics of PRGminer in Phase II (Eight-Class Classification)
| Metric | k-fold Training/Testing Performance | Independent Testing Performance |
|---|---|---|
| Overall Accuracy | 97.55% | 97.21% |
| Matthews Correlation Coefficient (MCC) | 0.93 | 0.92 |
The high overall accuracy and MCC values across all eight classes demonstrate that the model effectively distinguishes between the nuanced domain architectures of different R-gene types [3] [8]. This allows researchers to place high confidence in the specific class assignment.
Experimental Protocol for Phase II Validation
Objective: To validate the multi-class classification performance of PRGminer's Phase II model. Input: A set of protein sequences pre-validated as R-genes. Method:
- Input Transfer: Use the R-gene sequences identified in Phase I as input for Phase II.
- Model Loading: Load the pre-trained Phase II deep learning model.
- Classification Execution: Submit the R-gene sequences for detailed classification.
- Output Collection: For each sequence, record the predicted class (one of the eight categories) and the confidence score associated with that specific class.
- Performance Calculation: Compare the predicted classes against the known, domain-based annotations to calculate overall accuracy and per-class metrics.
The Scientist's Toolkit: Research Reagent Solutions
The following table details key resources used in the development and application of tools like PRGminer, which are essential for researchers in this field.
Table 3: Essential Research Reagents and Resources for R-gene Analysis
| Resource Name | Type | Function in R-gene Research |
|---|---|---|
| PRGminer Webserver | Web Tool | Freely accessible online platform for predicting and classifying R-genes from protein sequences [3]. |
| PRGminer Standalone | Software | Downloadable version of the tool for local installation and batch analysis [3] [8]. |
| PRGdb | Curated Database | A key repository of known and putative pathogen recognition genes (PRGs), supporting annotation and comparative analysis [30] [14]. |
| Phytozome | Genomic Database | Provides integrated genomic and functional data for plant genomes, used as a data source for training sets [3] [8]. |
| Ensembl Plants | Genomic Database | Another primary source of plant genome data and bio-mart tools for domain information retrieval [3] [8]. |
| NCBI Genome DB | Genomic Database | The National Center for Biotechnology Information database, a fundamental resource for sequence data [3] [8]. |
| InterProScan | Bioinformatics Tool | Used for protein domain analysis and functional prediction; a component of some traditional R-gene identification pipelines [14]. |
| BioVizSeq | R Package | A visualization tool for creating publication-quality figures of functional elements on biological sequences, which can be used to illustrate R-gene domain structures [31]. |
Best Practices for Result Application
- Confidence Thresholding: For high-stakes applications (e.g., selecting candidates for experimental validation), consider setting a minimum confidence score threshold (e.g., >0.95) to minimize false positives.
- Biological Plausibility: Always cross-reference predictions with biological context. For example, a high-confidence TNL prediction should be plausible within the known R-gene repertoire of the studied plant species.
- Sequence Quality Check: Ensure input protein sequences are high-quality and full-length, as fragmented sequences may lead to misclassification or low confidence scores due to missing domains.
- Multi-Tool Corroboration: For critical discoveries, consider validating PRGminer's predictions with other complementary tools and databases, such as PRGdb [30] or domain-based scanners, to strengthen the evidence.
Maximizing PRGminer Performance: Data and Workflow Best Practices
The accurate prediction of plant resistance (R-) genes using deep learning models is fundamentally dependent on the quality and proper formatting of input protein sequences. PRGminer, a state-of-the-art deep learning tool, exemplifies this principle, achieving remarkable prediction accuracy of up to 98.75% when provided with correctly formatted data [3]. This application note details the essential protocols for preparing protein sequence data to leverage PRGminer's two-phase prediction framework, which first identifies R-genes from non-R-genes and subsequently classifies them into specific structural categories [3]. Proper data preparation ensures that researchers can reliably harness this tool to accelerate the discovery of novel resistance genes, understand plant immunity mechanisms, and develop disease-resistant crop varieties through informed breeding strategies.
PRGminer Input Specifications and Sequence Requirements
PRGminer accepts protein sequences in standard FASTA format through multiple submission methods, providing flexibility for different research scenarios [9]. The tool's input handling system is designed to accommodate both single sequences for targeted analysis and batch processing for high-throughput studies.
Table 1: PRGminer Input Methods and Specifications
| Input Method | Format Requirements | Use Case Scenarios | Limitations |
|---|---|---|---|
| Accession ID | Valid NCBI or UniProt identifiers | Quick analysis of known proteins | Dependent on external database availability |
| FASTA Upload | Single or multiple sequences in FASTA format | Batch processing of novel sequences | File size constraints based on server capacity |
| Direct Paste | FASTA-formatted text in text area | Immediate analysis of individual sequences | Manual processing impractical for large datasets |
The system processes these inputs through a structured workflow where Phase I performs binary classification (R-gene vs. non-R-gene) using dipeptide composition features, achieving 95.72% accuracy on independent testing [3]. Sequences classified as R-genes then proceed to Phase II, where they are categorized into eight specific classes based on their domain architectures: CNL, TNL, TIR, RLK, RLP, LYK, LECRK, and KIN [3]. This hierarchical approach ensures comprehensive characterization of resistance gene candidates while maintaining high classification accuracy of 97.21% on independent test sets [3].
Experimental Protocols for Sequence Preparation and Validation
Protocol: Retrieving and Formatting Sequences from Public Databases
Purpose: To obtain properly formatted protein sequences for PRGminer analysis from public biological databases. Materials: Computer with internet access, list of protein accession IDs or genomic data. Duration: 30 minutes to several hours depending on dataset size.
Sequence Acquisition:
- Download protein sequences from curated databases including Phytozome, Ensemble Plants, or NCBI [3]
- For genomic data, perform initial gene prediction using standard annotation pipelines
- Extract putative protein sequences from predicted gene models
FASTA Formatting:
- Begin each entry with a ">" symbol followed by sequence identifier
- Use alphanumeric characters without special symbols (except underscores)
- Include unique sequence identifiers for traceability
- Enter protein sequence using standard one-letter amino acid codes
- Ensure sequences contain only valid amino acid characters (A-Z, except B, J, O, U, X, Z)
- Remove trailing spaces, line breaks within sequences, and non-standard characters
Quality Verification:
- Verify sequence lengths correspond to plausible protein lengths (>50 amino acids)
- Check for internal stop codons (represented as asterisks) which may indicate sequencing errors
- Confirm unique identifiers for each entry in multi-FASTA files
Protocol: Curating Training Datasets for Custom Model Development
Purpose: To create robust training datasets for developing specialized prediction models. Materials: Known R-gene sequences, non-R-gene sequences, computational resources for feature extraction. Duration: Several days to weeks depending on dataset complexity.
Data Collection:
- Compile positive dataset (R-genes) from validated sources with experimental evidence
- Assemble negative dataset (non-R-genes) from the same proteomes to ensure consistent background
- Maintain balanced representation between positive and negative examples
Feature Extraction:
- Compute dipeptide composition (400-dimensional feature vector) as this representation yielded optimal performance in PRGminer [3]
- Calculate occurrence frequencies of all possible dipeptide pairs (AA, AC, AD...YY, YV, YW)
- Normalize dipeptide frequencies to account for sequence length variations
- Optional: Extract additional features including amino acid composition, transition probabilities, and physicochemical properties
Dataset Partitioning:
- Implement k-fold cross-validation (k=5 or k=10) for model training and validation
- Reserve independent test set (20-30% of data) for final performance evaluation
- Ensure no significant sequence similarity between training and test sets to prevent overestimation
Workflow Visualization: Data Preparation to Prediction Pipeline
The following diagram illustrates the complete workflow from data preparation through prediction and classification in PRGminer:
Table 2: Key Research Reagent Solutions for R-gene Prediction Studies
| Resource Category | Specific Tools/Services | Function in R-gene Prediction | Implementation Notes |
|---|---|---|---|
| Sequence Databases | Phytozome, Ensemble Plants, NCBI Protein | Source of validated protein sequences for training and comparison | Curated databases ensure data quality and annotation reliability |
| Deep Learning Framework | PRGminer Web Server, Standalone Package | Core prediction engine for identifying and classifying R-genes | Dipeptide composition features yield 98.75% accuracy in k-fold validation [3] |
| Feature Extraction | Custom Python scripts for dipeptide computation | Transform protein sequences into numerical features for deep learning | 400-dimensional feature vectors representing all possible dipeptide combinations |
| Validation Resources | Experimental R-gene repositories | Benchmark prediction accuracy against biologically confirmed R-genes | Essential for calculating performance metrics (MCC: 0.91-0.98) [3] |
| Computational Infrastructure | Local installation (Python 3.7+), GPU acceleration | Enable processing of large datasets (>10,000 sequences) | Required for customized analysis workflows and pipeline integration [9] |
Proper data preparation and sequence formatting are foundational to obtaining reliable results from PRGminer and similar deep learning tools for plant resistance gene prediction. By adhering to the protocols outlined in this document, researchers can ensure their input data meets the quality standards necessary for optimal model performance. The demonstrated accuracy of PRGminer, achieving 95.72% in Phase I and 97.21% in Phase II classification on independent test sets [3], highlights the effectiveness of this approach when implemented with carefully prepared input data. As the field advances, these data preparation standards will facilitate more accurate discovery of resistance genes, ultimately contributing to enhanced crop protection and sustainable agriculture.
For researchers utilizing deep learning tools like PRGminer for the prediction of plant resistance genes (R-genes), the choice between a web server and local installation is a critical decision that impacts research efficiency, scalability, and data security [3]. PRGminer is a state-of-the-art deep learning tool that identifies and classifies plant resistance proteins through a two-phase process: initial prediction of input protein sequences as R-genes or non-R-genes, followed by classification of positive hits into one of eight distinct classes with high accuracy [3] [24]. This application note provides a structured framework to help researchers and bioinformaticians select the optimal deployment strategy based on their specific dataset characteristics and computational requirements.
Quantitative Comparison: Web Server vs. Local Installation
The decision between web server and local installation primarily hinges on the scale of data and specific research workflow requirements. The table below summarizes the key comparative factors:
Table 1: Decision Matrix for PRGminer Deployment Options
| Factor | Web Server | Local Installation |
|---|---|---|
| Recommended Dataset Size | Small to medium datasets (< 10,000 sequences) [9] | Large datasets (> 10,000 sequences) [9] |
| Performance & Speed | Subject to network latency and server queue times | Full control over computational resources; optimized for batch processing [9] |
| Data Privacy | Data transferred to external server | Data remains within institutional infrastructure [9] |
| Integration Capabilities | Limited to web interface functionalities | Can be integrated into existing bioinformatics pipelines [9] |
| Customization | Fixed parameters and analysis types | Customizable analysis workflows and parameters [9] |
| Internet Dependency | Required | Not required after installation [9] |
| Cost | Free access [3] | Requires institutional computational resources |
Workflow and Decision Pathway
The following diagram illustrates the recommended decision-making workflow for selecting between PRGminer's web server and local installation, incorporating key considerations from Table 1:
Experimental Protocols for Deployment and Validation
Protocol A: Utilizing the PRGminer Web Server
This protocol is designed for researchers with smaller datasets who prefer a user-friendly interface without installation overhead.
4.1.1 Input Preparation
- Obtain protein sequences in FASTA format from sources such as Phytozome, Ensemble Plants, or NCBI [3].
- For accession IDs, use NCBI or UniProt identifiers for direct fetching [9].
- Validate file integrity and format before submission.
4.1.2 Submission Process
- Access the PRGminer webserver at https://kaabil.net/prgminer/ [3].
- Select preferred input method: accession ID, FASTA file upload, or direct sequence paste [9].
- Click "Run Prediction" to initiate analysis.
- Monitor job status via the web interface.
4.1.3 Results Interpretation
- Access results through the web interface upon completion.
- Interpret prediction outcomes (R-gene or Non-R-gene) with associated confidence scores.
- Review detailed classification for R-genes into one of eight categories (CNL, TNL, etc.) [3].
- Download results in CSV, JSON, or FASTA format for further analysis [9].
Protocol B: Local Installation and Operation
This protocol provides instructions for installing and running PRGminer locally, suitable for large-scale analyses and pipeline integration.
4.2.1 System Requirements and Installation
- Ensure Python 3.7 or higher is installed on the system.
- Verify sufficient RAM capacity for large dataset processing [9].
- Install required dependencies from the provided requirements.txt file.
- Download the standalone tool from https://github.com/usubioinfo/PRGminer [3].
- Consider optional GPU support for accelerated processing [9].
4.2.2 Large Dataset Processing
- Implement data partitioning to manage memory usage effectively [32].
- Utilize batch processing capabilities for efficient computation.
- Monitor system resources during execution to prevent bottlenecks.
- Employ data quality checks and preprocessing to ensure input validity [32] [33].
4.2.3 Pipeline Integration
- Incorporate PRGminer into existing bioinformatics workflows using its API.
- Automate analysis pipelines using scripting capabilities.
- Generate custom output formats compatible with downstream analysis tools.
- Implement logging and error handling for robust automated operations.
Protocol C: Experimental Validation of PRGminer Predictions
This protocol outlines a methodology for validating PRGminer performance on custom datasets, ensuring reliable results for research purposes.
4.3.1 Performance Benchmarking
- Curate a validation set of known R-genes and non-R-genes with experimental verification.
- Execute PRGminer analysis on the validation set using either web server or local installation.
- Calculate standard performance metrics: accuracy, precision, recall, and Matthews Correlation Coefficient (MCC).
- Compare results with published performance (95.72% accuracy on independent testing for Phase I) [3] [24].
4.3.2 Cross-Validation Implementation
- Implement k-fold cross-validation to assess model stability and reduce overfitting [33].
- For large datasets, use learning curves to determine the optimal training set size [33].
- Evaluate variance in performance across different folds to identify potential issues.
- Compare cross-validation results with simple train-test splits to determine the most efficient validation approach [33].
Table 2: Key Research Reagents and Computational Resources for PRGminer Research
| Resource/Reagent | Function/Application | Source/Availability |
|---|---|---|
| Protein Sequence Data | Primary input for R-gene prediction; requires FASTA format | Phytozome, Ensemble Plants, NCBI [3] |
| Experimental Validation Set | Ground truth data for benchmarking tool performance | Literature-curated, experimentally verified R-genes |
| Computational Infrastructure | Hardware resources for local installation and large-scale analysis | Institutional HPC resources or high-memory workstations [9] |
| Data Cleaning Tools | Preprocessing and quality control of input datasets | Custom scripts or data management tools [32] |
| Benchmark Datasets | Standardized datasets for performance comparison and validation | Public repositories (e.g., Kaggle, UCI) [34] [35] |
| Pipeline Automation Tools | Scripting frameworks for workflow automation | Python, Snakemake, Nextflow |
The choice between PRGminer's web server and local installation is fundamentally determined by research scale and requirements. The web server offers accessibility and ease of use for smaller datasets and exploratory analyses, while local installation provides the computational power, flexibility, and data security essential for large-scale genomics research and pipeline integration. By following the structured decision pathway, experimental protocols, and utilizing the appropriate resources outlined in this application note, researchers can optimize their deployment strategy to efficiently advance plant resistance gene discovery and characterization.
In deep learning applications for genomic prediction, the relationship between overfitting and model confidence scores critically determines real-world utility. Within plant genomics, tools like PRGminer—a deep learning-based high-throughput resistance gene (R-gene) prediction tool—demonstrate how properly calibrated confidence scores can enhance breeding programs and disease resistance research [3]. PRGminer operates through a two-phase prediction framework: initial R-gene identification followed by classification into eight distinct molecular classes [3] [11]. Despite achieving reported accuracies exceeding 98.75% in k-fold validation [3], the translation of these performance metrics to diverse, unseen plant genomes depends on robust generalization prevention against overfitting. This application note examines the interplay between overfitting and confidence estimation within PRGminer's architecture, providing experimental protocols and analytical frameworks for researchers developing similar genomic prediction tools.
Theoretical Foundations
Overfitting in Deep Genomic Models
Deep learning models applied to genomic sequence data exhibit unique overfitting characteristics compared to traditional computer vision or natural language processing applications. The high dimensionality of protein sequences, coupled with limited experimentally validated training examples, creates conditions where models may memorize phylogenetic biases rather than learning generalizable resistance determinants. PRGminer processes input protein sequences through dipeptide composition representations, achieving Matthews correlation coefficients of 0.98 during training and 0.91 during independent testing [3]. This performance gap, while moderate, indicates potential overfitting that must be quantified through confidence score analysis.
Confidence Score Calibration
In classification tasks, confidence calibration ensures that predicted probabilities accurately reflect true correctness likelihoods. For PRGminer's two-phase architecture, miscalibrated confidence scores could propagate errors from initial R-gene identification (Phase I) to subsequent classification (Phase II), potentially misdirecting breeding programs. The dipeptide composition feature representation that yielded optimal performance in PRGminer [3] provides a stable foundation for confidence estimation, though domain shift between training and deployment data requires continuous monitoring.
Table 1: Performance Metrics for PRGminer's Two-Phase Prediction Architecture
| Phase | Description | k-fold Accuracy | Independent Test Accuracy | Matthews Correlation Coefficient |
|---|---|---|---|---|
| Phase I | R-gene vs. Non-R-gene Prediction | 98.75% | 95.72% | 0.98 (training) / 0.91 (testing) |
| Phase II | R-gene Classification | 97.55% | 97.21% | 0.93 (training) / 0.92 (testing) |
Experimental Protocols
Protocol: Cross-Validation with Confidence Tracking
Purpose: Evaluate overfitting while monitoring confidence score distributions across validation folds.
Materials:
- Protein sequence datasets from Phytozome, Ensemble Plants, and NCBI [3]
- Experimentally validated R-genes and non-R-genes
- PRGminer software (available via https://github.com/usubioinfo/PRGminer) [3]
Procedure:
- Data Partitioning: Implement stratified k-fold cross-validation (k=5-10) maintaining class balances across R-gene categories [3]
- Model Training: For each fold, train PRGminer using dipeptide composition features with the architecture yielding 98.75% accuracy [3]
- Confidence Recording: For each prediction, record maximum softmax probability and predicted class
- Binning Analysis: Group confidence scores into 10 equally-spaced bins (0.0-0.1, 0.1-0.2, ..., 0.9-1.0)
- Calibration Calculation: Within each bin, compute accuracy as the proportion of correct predictions
- Expected Calibration Error (ECE): Calculate weighted average of absolute differences between bin accuracy and bin confidence [3]
Visualization: Generate reliability diagram plotting bin accuracy against bin confidence, with perfect calibration represented by the diagonal.
Protocol: Out-of-Distribution Confidence Assessment
Purpose: Evaluate confidence score behavior when encountering evolutionarily distant plant species not represented in training data.
Materials:
- Reference R-gene datasets spanning diverse plant taxa (e.g., Solanaceae, Poaceae, Fabaceae)
- Genomic data from newly sequenced plant species
- Domain similarity quantification tools (e.g., MMseqs2, HMMER3) [3]
Procedure:
- Dataset Curation: Compile protein sequences from phylogenetically distinct species relative to training data
- Feature Extraction: Generate dipeptide composition representations matching PRGminer's input specifications [3]
- Prediction & Confidence Recording: Process out-of-distribution samples through trained PRGminer model
- Confidence Distribution Analysis: Compare confidence score distributions between in-distribution and out-of-distribution samples
- Selectivity Calculation: Compute area under the receiver operating characteristic curve (AUROC) for distinguishing correct vs. incorrect predictions using confidence scores
- Threshold Optimization: Identify confidence thresholds that maximize F1-score for deployment decisions
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Reagent/Tool | Function in Overfitting Analysis | Application in PRGminer Context |
|---|---|---|
| Dipeptide Composition Features | Numerical representation of protein sequences for deep learning input | PRGminer's optimal feature representation achieving 98.75% accuracy [3] |
| Experimentally Validated R-genes | Gold-standard dataset for model training and validation | Curated from public databases (Phytozome, Ensemble Plants, NCBI) for benchmarking [3] |
| Domain-Specific Cross-Validation | Phylogenetically-aware evaluation to detect biogeographic bias | Stratified sampling across plant taxa to assess generalization [3] |
| Expected Calibration Error (ECE) | Quantitative measure of confidence-accuracy alignment | Monitoring discrepancy between predicted probability and empirical accuracy [3] |
| Out-of-Distribution Datasets | Testing model performance on evolutionarily distant species | Assessing confidence score degradation across plant families [3] |
| PRGminer Web Server | Accessible interface for model deployment | Available at https://kaabil.net/prgminer/ for community use [11] |
Analytical Framework
Confidence-Based Performance Metrics
Beyond traditional accuracy measures, confidence-aware evaluation provides deeper insights into model reliability for decision support in breeding pipelines. The following metrics should be tracked across model versions and deployments:
- Selectivity (AUROC): Ability of confidence scores to rank correct predictions higher than incorrect ones
- Calibration Error: Quantitative measure of alignment between confidence scores and empirical accuracy
- Brier Score: Composite measure of both calibration and refinement
- Confidence Histograms: Visualization of score distributions for correct and incorrect predictions
Table 3: Confidence Metric Interpretation Guidelines
| Metric | Optimal Value | Acceptable Range | Indication of Overfitting |
|---|---|---|---|
| Expected Calibration Error | 0.0 | < 0.05 | > 0.10 with higher test error |
| Selectivity (AUROC) | 1.0 | > 0.85 | < 0.70 with high training accuracy |
| Brier Score | 0.0 | < 0.25 | Discrepancy > 0.15 between train/test |
| Confidence Spread | Balanced distribution | Moderate skew acceptable | Extreme bimodality or universal high confidence |
Mitigation Strategies for Overfitting
When confidence metrics indicate overfitting, several mitigation strategies align with PRGminer's architecture and plant genomics domain:
Feature Space Regularization: Apply dropout or L2 regularization to dipeptide composition inputs, which yielded optimal performance in PRGminer [3]
Data Augmentation: Generate synthetic protein sequences through biologically plausible mutations and recombination events
Domain-Adaptation Training: Fine-tune models on increasingly phylogenetically distant species to improve generalization
Architecture Simplification: Reduce model complexity when confidence-accuracy alignment degrades on validation data
Ensemble Methods: Combine predictions from multiple models trained with different initializations or feature subsets
In deep learning applications for plant resistance gene prediction, understanding the relationship between overfitting and confidence scores enables more reliable deployment in breeding programs. PRGminer demonstrates how high accuracy (98.75% in k-fold validation) must be accompanied by robust confidence estimation (MCC 0.91 in independent testing) for practical utility [3]. The protocols and analytical frameworks presented here provide actionable approaches for quantifying and improving confidence calibration, particularly important when extending predictions to evolutionarily distant plant species. Through rigorous confidence-aware evaluation, researchers can develop more trustworthy genomic prediction tools that effectively balance performance with generalization, accelerating crop improvement programs and sustainable agriculture initiatives.
The identification of plant resistance genes ((R)-genes) is a cornerstone of modern crop improvement programs, essential for developing cultivars with durable disease resistance. Traditional methods for (R)-gene discovery, often reliant on sequence similarity and domain-based searches, face significant limitations in scalability and sensitivity, particularly when dealing with novel genes exhibiting low sequence homology to known proteins [3] [14]. The advent of deep learning has transformed this landscape, enabling the prediction of (R)-genes based on complex, hierarchical sequence patterns that evade conventional bioinformatics tools.
PRGminer emerges as a state-of-the-art, deep learning-based tool specifically designed for high-throughput prediction and classification of plant resistance genes from protein sequences [3]. Its implementation marks a significant advancement in computational plant pathology. This application note provides detailed protocols for the seamless integration of PRGminer into established genomics and bioinformatics pipelines, empowering researchers to leverage its high-accuracy predictions for accelerated gene discovery and functional characterization.
PRGminer is implemented as a two-phase deep learning system for comprehensive (R)-gene analysis [3] [11].
- Phase I: (R)-gene Prediction. The tool first classifies an input protein sequence as an (R)-gene or a non-(R)-gene. This initial screening is critical for filtering out irrelevant sequences and focusing computational resources on genuine candidates.
- Phase II: (R)-gene Classification. Sequences identified as (R)-genes in Phase I are subsequently classified into one of eight major structural classes, providing immediate insight into their potential domain architecture and mechanism of action [3] [11].
The tool's performance is robust, achieving an accuracy of 98.75% in (k)-fold testing and 95.72% on an independent test set during Phase I, with a high Matthews Correlation Coefficient (MCC) of 0.91 for independent testing [3]. Phase II classification also maintains a high overall accuracy of 97.21% on independent data [3]. This high level of accuracy is attributed to the use of dipeptide composition for sequence representation, which allows the underlying convolutional neural network (CNN) to effectively capture complex sequence patterns relevant to resistance protein function [3] [14].
Table 1: Key Performance Metrics of PRGminer [3]
| Phase | Description | k-fold Testing Accuracy | Independent Testing Accuracy | Independent Testing MCC |
|---|---|---|---|---|
| Phase I | R-gene vs. Non-R-gene | 98.75% | 95.72% | 0.91 |
| Phase II | R-gene Classification | 97.55% | 97.21% | 0.92 |
Table 2: Classification of Plant Resistance Genes by PRGminer [3] [11]
| Class Acronym | Class Name | Key Domains and Characteristics |
|---|---|---|
| CNL | Coiled-coil-NBS-LRR | Coiled-coil, Nucleotide-binding site, Leucine-rich repeat |
| TNL | TIR-NBS-LRR | Toll/Interleukin-1 receptor, NBS, LRR |
| RLK | Receptor-like kinase | Extracellular LRR, Transmembrane, Intracellular kinase domain |
| RLP | Receptor-like protein | Extracellular LRR, Transmembrane, Short cytoplasmic tail (no kinase) |
| LECRK | Lectin receptor-like kinase | Lectin, Kinase, and Transmembrane domains |
| LYK | Lysin motif receptor kinase | Lysin Motif (LysM), Kinase, and Transmembrane domains |
| KIN | Kinase | Kinase domain involved in resistance |
| TIR | Toll-interleukin receptor | TIR domain only, lacks LRR or NBS domains |
Integration Protocols
Integrating PRGminer into existing workflows enhances their predictive power and reduces dependency on less sensitive homology-based methods. Below are detailed protocols for two common advanced scenarios.
Protocol 1: Genome-Wide (R)-gene Discovery and Annotation
This protocol outlines the process for identifying and annotating (R)-genes from a newly assembled plant genome, creating a comprehensive resistance gene catalog.
Experimental Workflow:
Diagram 1: Genome-wide R-gene discovery workflow.
Detailed Methodology:
Input Data Preparation: Begin with a high-quality whole-genome assembly. Generate a comprehensive set of protein-coding gene models using standard ab initio and evidence-based annotation pipelines (e.g., BRAKER, MAKER). Extract the predicted protein sequences in FASTA format. This set serves as the input for PRGminer [3].
PRGminer Execution (Phase I):
- Tool Access: PRGminer can be accessed via its web server for smaller datasets or installed as a standalone tool from its GitHub repository for large-scale, genome-wide analyses [3].
- Command (Standalone): For the standalone version, use the provided script to run Phase I prediction.
- Output: The output is a list of protein sequences classified as (R)-genes or non-(R)-genes. Non-(R)-genes are excluded from further (R)-gene-specific analysis.
PRGminer Execution (Phase II):
- Input: Use the (R)-gene sequences identified in Phase I.
- Command (Standalone):
- Output: This step produces the classification of each (R)-gene into one of the eight classes (e.g., CNL, TNL, RLK) [11].
Data Integration and Cataloging:
- Combine the classification results with the original gene annotations (e.g., GFF3 file). This enriches the annotation by adding a custom attribute, such as
PRGminer_class. - The final output is a comprehensive (R)-gene catalog, which can be used to determine the distribution, diversity, and genomic organization (e.g., clustering) of resistance genes in the species of interest [14].
- Combine the classification results with the original gene annotations (e.g., GFF3 file). This enriches the annotation by adding a custom attribute, such as
Protocol 2: Targeted Discovery from Wild Relatives via RNA-Seq
This protocol is designed for the targeted discovery of novel (R)-genes from wild crop relatives, where resistance is often found, by leveraging transcriptomic data.
Experimental Workflow:
Diagram 2: Targeted R-gene discovery from RNA-Seq data.
Detailed Methodology:
Transcriptome Sequencing and Assembly:
- Plant Material: Collect tissue from a wild crop relative under pathogen challenge (e.g., at early time points post-infection) to capture the active immune response.
- Library Preparation: Perform standard RNA extraction, library preparation, and sequence on an Illumina platform. For more complete gene models, consider PacBio Iso-Seq.
- Assembly: Since a reference genome may not be available, perform de novo transcriptome assembly using tools like Trinity or SOAPdenovo-Trans to reconstruct transcripts.
Protein Sequence Extraction: Predict open reading frames (ORFs) from the assembled transcripts using a tool like TransDecoder. The resulting protein sequence file is the input for PRGminer.
PRGminer Analysis: Execute both Phase I and Phase II of PRGminer as described in Protocol 1. The key advantage here is that PRGminer's deep learning model can identify (R)-genes without relying on a reference genome or high sequence similarity, making it ideal for exploring genetically diverse wild relatives [3].
Candidate Gene Prioritization and Validation:
- Prioritization: Filter results to focus on highly expressed transcripts (using RNA-Seq read counts) that are classified as (R)-genes. Pay special attention to classes known to be associated with the target pathogen (e.g., CNL/TNL for intracellular effectors).
- Functional Validation: Clone the full-length coding sequence of top candidates and express them in a susceptible cultivar via transgenic methods or use virus-induced gene silencing (VIGS) to knock down candidate genes in a resistant background to confirm function [36]. Advanced genome editing tools like CRISPR-Cas9 can also be used to knock out or swap alleles for final validation [37].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for R-gene Discovery and Validation
| Tool / Resource | Type | Function in Pipeline | Key Features |
|---|---|---|---|
| PRGminer | Software Tool | Core (R)-gene prediction and classification | Deep learning model; >95% accuracy; 8-class classification [3] |
| BRAPI-compliant DBs (BreedBase, Germinate) | Database | Centralized data management for phenotypic and genotypic data | Enables seamless data retrieval and integration for correlation analysis [38] |
| QBMS 2.0 (R package) | Analytics Pipeline | Streamlines access to breeding data from multiple systems | Facilitates integration of field, pedigree, and genotyping data for analysis [38] |
| AlphaFold | Software Tool | Protein structure prediction | Predicts 3D structure of candidate R-proteins for functional analysis [36] |
| CRISPR-Cas9 | Molecular Biology | Functional validation of candidate (R)-genes | Enables precise gene editing to confirm gene function in plant immunity [36] [37] |
Concluding Remarks
The integration of PRGminer into genomic pipelines represents a significant leap forward for plant breeding and research. Its ability to accurately identify and classify resistance genes from sequence data alone, overcoming limitations of homology-based methods, allows researchers to efficiently tap into the rich diversity of (R)-genes, particularly in underutilized wild germplasm. The protocols outlined here provide a concrete roadmap for leveraging this powerful tool, from genome-wide annotation to targeted discovery. As the field moves toward Breeding 4.0, the synergy between deep learning tools like PRGminer, data interoperability standards like BrAPI, and advanced gene editing technologies will be instrumental in accelerating the development of disease-resistant crops, thereby enhancing global food security [25] [38] [37].
Benchmarking PRGminer: Accuracy, Limitations, and Future Directions
In the field of plant genomics, the accurate identification of resistance genes ((R)-genes) is a critical component for understanding plant defense mechanisms and guiding disease-resistant crop breeding programs. The PRGminer tool represents a significant advancement in this area, employing a deep learning-based approach to achieve high-throughput prediction of (R)-genes. This application note provides a detailed analysis of PRGminer's performance metrics, with particular focus on its exceptional performance in independent testing, where it achieved an accuracy exceeding 95% [3] [8]. We examine the experimental protocols, data composition, and computational methodologies that underpin these results, providing researchers with a comprehensive resource for implementing and validating this tool in their own workflows.
Experimental Design and Dataset Composition
The robustness of any predictive model is fundamentally dependent on the quality and composition of its training data. PRGminer was developed using protein sequence datasets sourced from major public databases including Phytozome, Ensemble Plants, and NCBI [3] [8]. To ensure data integrity and minimize redundancy, the researchers applied CD-HIT to eliminate duplicate sequences, then implemented a rigorous domain-based filtration system to distinguish true (R)-genes from non-(R)-genes.
Data Partitioning Strategy
A critical aspect of PRGminer's validation was the strategic partitioning of data into training and independent testing sets, which allows for unbiased evaluation of model performance on previously unseen data.
Table 1: Dataset Composition for Model Development and Validation
| Phase | Dataset Type | R-genes | Non-Rgenes | Total Sequences | Partition Ratio |
|---|---|---|---|---|---|
| Phase I | Initial Dataset | 18,952 | 19,212 | 38,164 | - |
| Phase I | Training/Validation (k-fold) | 17,057 | 17,291 | 34,348 | 90% |
| Phase I | Independent Testing | 1,895 | 1,921 | 3,816 | 10% |
| Phase II | R-gene Classification | 18,952 (across 8 classes) | - | 18,952 | 90% training, 10% testing |
For Phase I (R-gene vs. non-R-gene classification), the overall dataset of 18,952 (R)-genes and 19,212 non-Rgenes was divided in a 9:1 ratio, with 90% utilized for k-fold training and validation, and the remaining 10% held out as a completely independent dataset for final benchmarking [8]. This independent set was not used in any aspect of model training or hyperparameter tuning, ensuring an unbiased assessment of generalizability to novel sequences.
R-gene Classification Schema
Phase II of PRGminer involves the classification of predicted (R)-genes into specific functional categories based on their domain architecture [3] [8]. The eight classes represent major (R)-gene types with distinct roles in plant immunity:
- CNL: Coiled-coil, Nucleotide-binding site, Leucine-rich repeat domains
- KIN: Kinase domain
- RLP: Leucine-rich repeat and Transmembrane domains with a cytoplasmic region
- LECRK: Lectin, Kinase, and Transmembrane domains
- RLK: Extracellular Leucine-rich repeat and Kinase domains
- LYK: LysM domain, Kinase, and Transmembrane domains
- TIR: Toll/interleukin-1 receptor domain
- TNL: Toll/interleukin-1 receptor, Nucleotide-binding site, and Leucine-rich repeat domains
Performance Metrics and Validation Results
PRGminer was evaluated using multiple performance metrics to provide a comprehensive assessment of its predictive capabilities. The results demonstrate exceptional performance in both phases of prediction.
Phase I Performance: R-gene Identification
The initial classification phase distinguishes (R)-genes from non-Rgenes using dipeptide composition features fed into a deep learning architecture.
Table 2: Performance Metrics for Phase I (R-gene vs. Non-R-gene Prediction)
| Evaluation Type | Accuracy | Matthews Correlation Coefficient (MCC) | Key Feature Representation |
|---|---|---|---|
| k-fold Training/Testing | 98.75% | 0.98 | Dipeptide Composition |
| Independent Testing | 95.72% | 0.91 | Dipeptide Composition |
The high Matthews Correlation Coefficient (MCC) values are particularly noteworthy, as this metric provides a more reliable measure of binary classification performance than accuracy alone, especially when dealing with imbalanced datasets. An MCC of 0.91 on independent testing indicates strong agreement between predicted and actual classifications [3] [8].
Phase II Performance: R-gene Classification
For sequences identified as (R)-genes in Phase I, Phase II performs fine-grained classification into one of the eight categories based on domain architecture.
Table 3: Performance Metrics for Phase II (R-gene Classification)
| Evaluation Type | Overall Accuracy | Matthews Correlation Coefficient (MCC) |
|---|---|---|
| k-fold Training/Testing | 97.55% | 0.93 |
| Independent Testing | 97.21% | 0.92 |
The maintained high accuracy and MCC in independent testing for Phase II demonstrates that the model effectively learns the discriminative features between different (R)-gene classes without overfitting to the training data [8].
Computational Methodology and Workflow
PRGminer implements a sophisticated deep learning approach that extracts both sequential and convolutional features directly from raw encoded protein sequences, moving beyond traditional alignment-based methods that struggle with low-homology sequences [3] [8].
PRGminer Two-Phase Prediction Workflow
The following diagram illustrates the complete predictive workflow implemented in PRGminer, from input to final classification:
Deep Learning Architecture
PRGminer utilizes a convolutional neural network (CNN) architecture capable of extracting both local and global features from protein sequences. The model processes dipeptide composition representations of sequences, which provided superior performance compared to other feature representation methods [3].
To facilitate the implementation of PRGminer in research workflows, the following table details key computational resources and their functions in the prediction process.
Table 4: Essential Research Reagents and Computational Resources
| Resource Name | Type | Function in PRGminer Workflow | Access Information |
|---|---|---|---|
| PRGminer Webserver | Web Application | Primary interface for sequence prediction | Freely accessible at: https://kaabil.net/prgminer/ [3] |
| PRGminer Standalone | Software Package | Local installation for large-scale analysis | Download: https://github.com/usubioinfo/PRGminer [3] |
| Dipeptide Composition | Feature Encoding Method | Numerical representation of protein sequences for deep learning | Implementation details in publication [3] [8] |
| Phytozome | Data Source | Source of curated plant protein sequences for training | Public database [8] |
| Ensemble Plants | Data Source | Source of annotated plant genomes for training | Public database [8] |
| NCBI Protein Database | Data Source | Comprehensive repository of protein sequences | Public database [3] [8] |
| CD-HIT | Bioinformatics Tool | Removal of redundant sequences from training data | Standard tool for sequence redundancy reduction [8] |
Step-by-Step Experimental Protocol
Protocol 1: Web-Based Prediction Using PRGminer
This protocol describes the procedure for predicting (R)-genes using the PRGminer webserver, suitable for analyzing individual sequences or small batches.
- Input Preparation: Prepare your protein sequence(s) in FASTA format. Alternatively, note relevant accession IDs from NCBI or UniProt databases.
- Sequence Submission: Access the PRGminer webserver at https://kaabil.net/prgminer/ and submit sequences using one of three methods [9]:
- Accession ID: Enter a valid protein accession ID (e.g., from NCBI) in the designated field.
- FASTA Upload: Use the file upload feature to submit a FASTA file containing one or multiple protein sequences.
- Direct Paste: Manually paste FASTA-formatted sequence data directly into the input text area.
- Initiate Prediction: Click the "Run Prediction" button to initiate the analysis. The system will automatically process the sequence through both Phase I and Phase II classifiers.
- Results Interpretation: After processing, review the results table which includes [9]:
- Sequence identification information
- Phase I prediction outcome (R-gene or Non-R-gene)
- Confidence scores for the prediction
- For sequences classified as R-genes: Detailed Phase II classification into one of the eight categories
- Results Download: Download complete results in CSV, JSON, or FASTA format for further analysis. Filtered downloads based on specific R-gene classes or confidence thresholds are also available.
Protocol 2: Large-Scale Analysis Using Standalone Package
For researchers working with large datasets (>10,000 sequences) or requiring integration with existing pipelines, local installation is recommended [9].
System Requirements Verification: Ensure the host system meets the following requirements:
- Python 3.7 or higher
- Sufficient RAM for large datasets (16GB minimum recommended for large-scale analyses)
- GPU support (optional but recommended for accelerated processing)
Software Installation:
- Download the standalone package from https://github.com/usubioinfo/PRGminer
- Install required dependencies using the provided requirements.txt file
- Verify successful installation by running basic tests
Batch Processing:
- Prepare input files in FASTA format
- Configure batch processing parameters as needed
- Execute the prediction pipeline on the entire dataset
- Monitor processing progress for large jobs
Output Analysis:
- Process output files for integration with downstream analyses
- Apply custom filtering based on confidence thresholds
- Generate summary statistics for large-scale predictions
The performance metrics demonstrate that PRGminer represents a significant advancement in (R)-gene prediction technology. The achievement of >95% accuracy on independent testing, coupled with high Matthews Correlation Coefficients across both classification phases, indicates a model with strong predictive power and excellent generalizability to novel sequences [3] [8]. This performance substantially outperforms traditional alignment-based methods such as BLAST, InterProScan, and HMMER, particularly for sequences with low homology to previously characterized (R)-genes [8].
The two-phase classification approach allows researchers to not only identify potential (R)-genes but also gain immediate insights into their likely functional classification based on domain architecture. This dual-level information is particularly valuable for prioritizing candidates for further experimental validation in breeding programs.
The availability of both web-based and standalone versions makes PRGminer accessible to researchers with varying computational resources and expertise levels. The web server offers convenience for individual sequence analysis, while the standalone package supports large-scale genomic analyses and integration with existing bioinformatics pipelines [9].
As plant genomic data continues to expand at an accelerating pace, tools like PRGminer that leverage deep learning for functional annotation will play an increasingly vital role in translating sequence information into biological insights. The high-accuracy performance documented in this analysis positions PRGminer as a valuable resource for the plant research community, with potential applications in fundamental research on plant immunity and applied crop improvement strategies aimed at enhancing disease resistance.
In the field of plant genomics, the accurate prediction of plant resistance genes (R-genes) is crucial for understanding plant defense mechanisms and guiding disease resistance breeding programs. Researchers have traditionally relied on alignment-based tools and traditional machine learning (ML) methods for this task. Recently, however, deep learning (DL) approaches have emerged as powerful alternatives [3] [39]. This application note provides a detailed, head-to-head comparison of these methodologies, using the deep learning-based PRGminer tool as a central case study within a broader thesis on deep learning prediction of plant resistance genes [3]. We present structured quantitative comparisons, detailed experimental protocols, and essential resource toolkits to guide researchers in selecting and implementing the most appropriate method for their specific R-gene prediction projects.
Performance Comparison: Quantitative Analysis
The table below summarizes the core performance characteristics of alignment-based, traditional machine learning, and deep learning methods for plant resistance protein prediction, synthesizing data from recent studies.
Table 1: Comparative Performance of R-gene Prediction Methods
| Feature | Alignment-Based Methods | Traditional Machine Learning | Deep Learning (e.g., PRGminer) |
|---|---|---|---|
| Theoretical Basis | Sequence similarity, motif search [3] [39] | Statistical models, hand-crafted features (e.g., composition) [39] [40] | Multi-layered neural networks for automated feature extraction [41] [3] |
| Representative Tools | BLAST, HMMER, InterProScan [3] [39] | SVM-based models [39] [42] | PRGminer, CNNs [3] [42] |
| Reported Accuracy (AUROC/Acc) | Poor coverage/sensitivity [39] | AUROC: 0.91-0.95 [39] | Accuracy: 98.75% (k-fold), 95.72% (independent test) [3] |
| Key Strength | Simplicity, well-established | Effective with smaller, structured datasets; more interpretable [43] [44] | High accuracy with complex data; automated feature learning [41] [3] |
| Key Limitation | Fails with low homology, poor sensitivity [3] [39] | Requires manual feature engineering [43] [44] | High computational cost; "black box" model [43] [44] |
| Data Dependency | Reference databases | Smaller, structured datasets [44] [45] | Large volumes of data (e.g., thousands of data points per feature) [43] [3] |
| Hardware Requirement | Standard computers | Standard computers [45] | High-performance GPUs/TPUs [43] [45] |
Experimental Protocols for Method Evaluation
Protocol for Alignment-Based R-gene Prediction
This protocol outlines the use of common alignment-based tools for identifying resistance genes, a method known for its simplicity but limited sensitivity with sequences of low homology [3] [39].
- Input Data Preparation: Compile protein or nucleotide sequences of interest in FASTA format.
- Sequence Database Search:
- Domain and Motif Identification:
- Result Integration and Annotation: Manually curate and combine the results from the similarity search and domain analysis to assign a putative R-gene classification.
Protocol for Traditional Machine Learning-Based Prediction
This protocol describes the workflow for building a predictive model using traditional ML, which depends on effective feature engineering [39] [42].
- Dataset Curation:
- Positive Data: Collect confirmed R-protein sequences from public databases like Phytozome and Ensemble Plants [3].
- Negative Data: Assemble a set of non-R-protein sequences from the same sources.
- Partitioning: Split the dataset into training, validation, and independent testing sets, ensuring no protein in the test set exceeds 40% similarity to those in the training set to rigorously assess generalizability [39].
- Feature Engineering:
- Feature Extraction: Compute numerical features from the raw protein sequences. Common features include:
- Feature Selection: Apply feature selection algorithms to reduce dimensionality and mitigate overfitting.
- Model Training and Validation:
- Model Evaluation: Use the held-out independent test set to evaluate the final model's performance, reporting metrics like accuracy, sensitivity, specificity, and Area Under the ROC Curve (AUROC) [39].
Protocol for Deep Learning-Based Prediction with PRGminer
This protocol details the specific two-phase workflow of the PRGminer tool, which leverages deep learning to automate feature extraction and achieve high prediction accuracy [3].
- Data Acquisition and Preprocessing:
- Phase I - R-gene Identification:
- Objective: Classify the input protein sequence as either an R-gene or a non-R-gene.
- Model Architecture: A deep learning model (specific architecture not detailed in the search results) is trained on the encoded sequences.
- Performance Benchmark: PRGminer achieved an accuracy of 98.75% in k-fold testing and 95.72% on an independent test set in this phase [3].
- Phase II - R-gene Classification:
- Objective: Classify sequences identified as R-genes in Phase I into one of eight specific classes.
- Classes: CNL, TNL, TIR, RLK, RLP, LECRK, LYK, and KIN [3].
- Performance Benchmark: PRGminer achieved an overall accuracy of 97.55% in k-fold testing and 97.21% on an independent test set in this classification phase [3].
- Output and Analysis: The tool provides predictions and classifications, which should be reviewed by researchers for downstream application.
Diagram 1: PRGminer's two-phase DL workflow for R-gene identification and classification.
The Scientist's Toolkit: Research Reagent Solutions
The table below lists key resources for developing and implementing R-gene prediction models.
Table 2: Essential Research Reagents and Resources for R-gene Prediction
| Resource Name | Type | Primary Function in R-gene Research |
|---|---|---|
| PRGminer | Deep Learning Tool | A specialized DL-based webserver and standalone tool for high-throughput identification and classification of plant R-genes [3]. |
| PlantDRPpred | Machine Learning Tool | An online platform that uses an ensemble ML model for predicting and designing plant disease resistance proteins [39]. |
| Phytozome | Genomic Database | A key public database for plant genomic data, serving as a primary source for obtaining R-gene and non-R-gene sequences for model training [3]. |
| NCBI Database | Genomic Database | A comprehensive repository of protein and nucleotide sequences, used for gathering experimental data and building reference sets [3] [39]. |
| BLAST Suite | Alignment-Based Tool | The standard tool for performing initial sequence similarity searches against known R-gene databases [3] [39]. |
| InterProScan | Domain Analysis Tool | A software suite used for functional analysis of proteins by classifying them into families and predicting domains and motifs [3]. |
| Dipeptide Composition | Feature Encoding Method | A simple numerical representation of protein sequences that effectively captures compositional information for ML/DL models [3]. |
Workflow Comparison and Decision Framework
The following diagram summarizes the logical relationship and fundamental differences in the workflows of the three compared methodologies, highlighting the reduced need for manual intervention in the deep learning approach.
Diagram 2: A simplified comparison of the core workflows for alignment-based, traditional ML, and deep learning methods.
The identification of resistance genes (R-genes) in crop plants is a critical research focus for sustainable agriculture, aimed at reducing reliance on chemical pesticides. Plant R-genes encode proteins that detect specific pathogen effectors, initiating a powerful immune response known as effector-triggered immunity (ETI) [3]. Over the past three decades, more than 450 R-genes have been cloned from 42 plant species, with about 72% encoding cell surface or intracellular NLR immune receptors [46]. Traditional cloning methods are increasingly being supplemented by advanced genomic approaches, including Genome-Wide Association Studies (GWAS) and deep learning-based prediction tools. These innovations are accelerating the discovery and deployment of R-genes in major crops. This article explores successful case studies of genome-wide R-gene identification, with a specific focus on the integration of the deep learning tool PRGminer into the research pipeline, and provides detailed protocols for its application.
The PRGminer Tool: A Deep Learning Framework for R-gene Prediction
PRGminer is a state-of-the-art, deep learning-based bioinformatic tool specifically designed for the high-throughput identification and classification of plant resistance genes [3]. Its development addresses significant challenges in R-gene discovery, such as their complex genomic structure, low expression levels, and the limitations of homology-based prediction methods, which often fail when sequence homology is low [3].
The tool operates through a streamlined, two-phase analytical workflow (Figure 1), achieving exceptional accuracy in both phases.
Figure 1. PRGminer's two-phase analysis workflow. The tool first predicts whether an input protein is a resistance gene, then classifies positive hits into one of eight specific classes.
- Phase I: R-gene Prediction. In this initial phase, PRGminer analyzes input protein sequences to classify them as either R-genes or non-R-genes. Among various sequence representations tested, the dipeptide composition achieved the best prediction performance, with an accuracy of 98.75% in a k-fold training/testing procedure and 95.72% on independent testing [3].
- Phase II: R-gene Classification. Sequences identified as R-genes in Phase I are subsequently classified into one of eight major structural classes. This phase achieves an overall accuracy of 97.55% (k-fold) and 97.21% on independent testing [3].
The high accuracy and automated nature of PRGminer make it an invaluable resource for researchers aiming to accelerate the discovery of new R genes, understand the genetic basis of plant resistance, and develop strategies for breeding resistant crops [3] [11].
Table 1: Performance Metrics of PRGminer
| Phase | Description | k-fold Testing Accuracy | Independent Testing Accuracy | MCC Value |
|---|---|---|---|---|
| Phase I | R-gene vs. Non-R-gene Prediction | 98.75% | 95.72% | 0.98 (k-fold), 0.91 (independent) |
| Phase II | R-gene Classification | 97.55% | 97.21% | 0.93 (k-fold), 0.92 (independent) |
Table 2: The Eight R-gene Classes Identified by PRGminer
| Class | Name | Key Domains and Features |
|---|---|---|
| CNL | Coiled-coil-NBS-LRR | Coiled-coil (CC), Nucleotide-binding site (NBS), Leucine-rich repeat (LRR) [3]. |
| TNL | TIR-NBS-LRR | Toll/Interleukin-1 receptor (TIR), NBS, LRR [3]. |
| RLK | Receptor-like kinase | Extracellular leucine-rich repeat (eLRR), Kinase domain [11]. |
| RLP | Receptor-like protein | eLRR, Transmembrane region, Short cytoplasmic tail (no kinase domain) [11]. |
| LECRK | Lectin receptor-like kinase | Lectin motif (LECM), Kinase, Transmembrane (TM) domain [3]. |
| LYK | Lysin motif receptor kinase | Lysin Motif (LYSM), Kinase, TM domain [3]. |
| KIN | Kinase | Kinase domain involved in the resistance process [11]. |
| TIR | Toll-interleukin receptor | TIR domain only, lacks LRR or NBS domains [11]. |
Case Study 1: Identification of a Rice Stem Borer Resistance Gene
Background and Objective
The striped stem borer (SSB) is a major Lepidopteran pest that causes significant yield losses in rice. As chemical control is problematic, identifying endogenous resistance genes is a priority. A research group aimed to identify quantitative trait loci (QTLs) and candidate genes conferring SSB resistance in a diverse panel of 201 rice cultivars [47].
Integrated Experimental Protocol
Step 1: Phenotypic Evaluation of Resistance
- Plant Materials: 201 rice cultivars from diverse geographical origins were cultivated in field conditions [47].
- Insect Inoculation: At the peak tillering stage, first-instar SSB larvae ("ant borers") were manually inoculated onto rice plants. The number of larvae applied was half the number of tillers per plant [47].
- Damage Assessment: Thirty days post-inoculation, the number of withered hearts and tillers was counted. The corrected damage index (D) was calculated as the ratio of the damage index of the test plant to that of a susceptible control (TN1) [47].
Step 2: Genotyping and Genome-Wide Association Study (GWAS)
- Genotyping: The 201 rice samples were genotyped using 2,849,855 high-confidence single nucleotide polymorphisms (SNPs) [47].
- GWAS Analysis: Association analysis between SNP markers and the corrected damage index was performed using a mixed linear model to account for population structure and kinship [47].
Step 3: Candidate Gene Identification and Functional Validation
- QTL and Gene Annotation: The GWAS identified a major QTL,
qRSSB4. Gene annotation within this locus, combined with qRT-PCR expression analysis in resistant cultivars, prioritizedLOC_Os04g34140(namedOsRSSB4) as a candidate gene [47]. - Transgenic Validation: The candidate gene
OsRSSB4was overexpressed in the susceptible variety Nipponbare to generate transgenic lines (OsRSSB4OE) [47]. - Bioassay Confirmation: Insect resistance of the overexpression lines was evaluated against the wild-type control. Results showed a dramatic reduction in the withering heart rate of transgenic lines (0-8.3%) compared to the wild-type (100%), confirming that
OsRSSB4positively regulates defense against SSB [47].
Case Study 2: Genome-Wide Discovery of Herbicide Resistance Genes in Rice
Background and Objective
Weeds pose a severe threat to rice yields. This study sought to identify novel endogenous genes conferring resistance to three commonly used herbicides (glufosinate, glyphosate, and mesotrione) by leveraging natural variation within a diverse rice population [48].
Integrated Experimental Protocol
Step 1: High-Throughput Phenotyping
- Plant Materials: A panel of 421 diverse cultivated rice varieties was used [48].
- Herbicide Treatment: At the 3-4 leaf stage, plants were sprayed with half-lethal doses of the three herbicides. Each variety was replicated three times [48].
- Injury Scoring: Herbicide injury was scored on a 0-4 scale five days (glyphosate and glufosinate) or seven days (mesotrione) after application [48].
Step 2: Multi-Model GWAS and Haplotype Analysis
- Genotyping and GWAS: The panel was genotyped with 6.3 million variants (SNPs and InDels). GWAS was performed for the entire panel and for indica and japonica subpopulations separately, using a mixed linear model [48].
- Variant Function Annotation: Significant associations were identified. The functional impact of polymorphisms within a 1 Mb region of associated loci was annotated using
snpEffsoftware. Priority was given to variants predicted to cause amino acid changes or alter splicing [48]. - Haplotype Analysis: The geographic distribution of resistant haplotypes was analyzed, revealing that favorable alleles for two major QTLs (
RGlu6andRGly8) were predominantly present in japonica cultivars from Europe [48].
A Standardized Protocol for R-gene Identification Integrating PRGminer
The following protocol outlines a streamlined workflow for genome-wide R-gene discovery, incorporating GWAS and the deep learning tool PRGminer.
Protocol: Genome-Wide Identification and Validation of R-genes in Crops
A. Preliminary Analysis and Candidate Prioritization
- Phenotyping: Conduct replicated trials to score disease or pest resistance in a diverse germplasm panel.
- Genotyping & GWAS: Perform high-density SNP genotyping followed by GWAS to identify significant marker-trait associations and define QTL regions.
- Candidate Gene Mining: Annotate all genes within the significant QTL intervals from a reference genome.
- In silico Screening with PRGminer:
- Input: Extract the protein sequences of all annotated genes within the QTL region.
- Processing: Submit the protein sequences to the PRGminer webserver (https://kaabil.net/prgminer/) or run the standalone tool (https://github.com/usubioinfo/PRGminer).
- Output Analysis: Prioritize genes that PRGminer predicts with high confidence as R-genes (Phase I) for further analysis. Use the Phase II classification to inform hypotheses about the potential mechanism of the resistance protein (e.g., CNL, RLK).
B. Functional Validation
- Expression Profiling: Validate the expression patterns of candidate genes (especially those prioritized by PRGminer) in resistant vs. susceptible lines under pathogen/herbicide challenge via qRT-PCR.
- Transgenic Validation: Conduct functional studies by overexpressing the candidate gene in a susceptible cultivar or knocking it down/out in a resistant cultivar, followed by rigorous phenotyping to confirm the gene's role in resistance.
The integration of PRGminer into this workflow provides a powerful filter, leveraging deep learning to add a functional prediction layer to positional cloning, thereby increasing the efficiency of candidate gene selection.
Table 3: Key Research Reagent Solutions for R-gene Identification
| Reagent / Resource | Function in Research | Example Application |
|---|---|---|
| Diverse Germplasm Panel | Provides the natural genetic variation needed to detect associations between genotype and phenotype. | 201 rice cultivars for SSB resistance [47]; 421 rice varieties for herbicide resistance [48]. |
| High-Density SNP Markers | Serve as genetic landmarks for GWAS to pinpoint genomic regions associated with the resistance trait. | 2.8 million SNPs for SSB GWAS [47]; 6.3 million variants for herbicide GWAS [48]. |
| PRGminer Webserver | Deep learning tool for accurate prediction and classification of R-genes from protein sequences. | Screening candidate genes within a QTL to prioritize those with structural hallmarks of known R-genes [3] [11]. |
| Transgenic Lines (Overexpression/KO) | Provides direct evidence for gene function by altering its expression and observing changes in the resistant phenotype. | OsRSSB4OE lines confirmed gene function in SSB resistance [47]. |
| Reference Genome Annotation | Provides the positional and functional context of genes within a defined QTL region. | Rice Annotation Project version 7.0 (Nipponbare) used for candidate gene identification [48]. |
The case studies presented demonstrate a powerful paradigm for R-gene discovery in crops. The integration of high-throughput genotyping, GWAS, and robust phenotyping successfully identified key loci for insect and herbicide resistance in rice. The incorporation of advanced bioinformatic tools like PRGminer further strengthens this pipeline by providing a fast, accurate, and deep learning-based method to screen and classify candidate genes, moving beyond reliance on sequence homology alone. These integrated genomic approaches, complemented by rigorous functional validation, are accelerating the development of disease- and pest-resistant crop varieties, which is fundamental to ensuring global food security.
The deployment of deep learning tools like PRGminer for the prediction of plant resistance (R-) genes represents a significant advancement in the field of plant bioinformatics [3]. These tools are crucial for accelerating the breeding of disease-resistant crops, a key component in safeguarding global food security [14]. However, the practical application of these models is confronted by several interconnected challenges: the inherent data scarcity of experimentally validated R-genes, the interpretability of complex deep learning predictions, and the continuous evolution of pathogens that can overcome plant resistance. This document provides detailed application notes and protocols, framed within the context of PRGminer research, to help researchers navigate these challenges effectively. By implementing robust data strategies, leveraging explainable AI techniques, and adopting evolutionary-aware validation protocols, scientists can enhance the reliability and impact of their computational predictions in both basic research and applied crop development.
Application Note: Overcoming Data Scarcity in R-gene Prediction
The development of robust deep learning models for R-gene discovery is fundamentally constrained by the limited availability of high-quality, curated training data. The following table summarizes the scale of data utilized by a state-of-the-art tool and the current landscape of cloned R-genes, illustrating the data scarcity problem.
Table 1: Data Resources for R-gene Prediction
| Data Resource / Tool | Reported Dataset Size / Availability | Key Features / Description | Performance Metrics |
|---|---|---|---|
| PRGminer Training Data [3] | Compiled from public databases (Phytozome, Ensemble Plants, NCBI) | Used for two-phase deep learning model; dipeptide composition feature representation | Phase I Accuracy: 98.75% (k-fold), 95.72% (independent test); Phase II Accuracy: 97.55% (k-fold), 97.21% (independent test) |
| Cloned R-genes in Major Crops [14] | >450 genes cloned across all plant species; ~460 documented in bread wheat; 46 in rice against bacterial blight | Provides a core set of experimentally validated sequences for model training and testing | Foundation for understanding domain architecture and resistance mechanisms |
Experimental Protocol: Data Augmentation and Model Training
This protocol outlines a strategy to mitigate data scarcity for training a custom R-gene prediction model, extending beyond the PRGminer framework.
Objective: To augment a limited set of known R-genes and train a high-performance predictive model. Materials: A small, curated set of experimentally validated R-gene sequences (e.g., from PRGdb or RefPlantNLR); a large, general plant proteome (e.g., from Phytozome or NCBI) to serve as a source of negative examples and for data augmentation.
Initial Data Curation:
- Positive Set Curation: Collect a core set of known R-genes from curated databases. Manually review literature to verify their domain architectures (e.g., CNL, TNL, RLK).
- Negative Set Curation: Extract protein sequences from plant proteomes that lack known R-gene domains (via InterProScan or HMMER3). Filter out any sequences with known NBS, LRR, TIR, or RLK domains to create a robust negative dataset.
Data Augmentation via Sequence Manipulation:
- Employ bioinformatics scripts (e.g., in Biopython) to generate synthetic R-gene variants.
- Techniques:
- Point Mutations: Introduce random, biologically plausible amino acid substitutions at a low rate (e.g., 1-2% of sequence length).
- In-frame Insertions/Deletions: Simulate the evolution of variable regions, particularly within the LRR domains, by making small, in-frame indels.
- Domain Shuffling: For sequences with multiple domains, create chimeras by swapping homologous domains between different R-genes from the same class.
Feature Engineering and Model Training:
- Feature Extraction: Following the PRGminer precedent, compute dipeptide composition for all sequences (original and augmented). This captures local sequence order information and has proven highly effective [3].
- Model Building:
- Partition the augmented dataset into training (80%), validation (10%), and hold-out test (10%) sets, ensuring no data leakage between splits.
- Train a deep learning classifier, such as a Multi-Layer Perceptron (MLP) or a 1D Convolutional Neural Network (CNN), using the validation set for hyperparameter tuning and early stopping.
- Performance Assessment: Finally, evaluate the model on the held-out test set using accuracy, Matthews Correlation Coefficient (MCC), and per-class F1 scores to ensure balanced performance across R-gene classes.
Application Note: Enhancing Model Interpretability
Deep learning models are often perceived as "black boxes," which limits trust and hinders the extraction of novel biological insights from their predictions [49]. Interpretability is not merely about explaining a model's decision but about enabling researchers to learn from the model to guide future experiments [50]. The following table categorizes key approaches to this challenge.
Table 2: Strategies for Interpreting R-gene Prediction Models
| Interpretability Strategy | Category | Description | Application in R-gene Prediction |
|---|---|---|---|
| SHAP (SHapley Additive exPlanations) [49] | Post-hoc, Model-agnostic | Assigns each feature (e.g., a dipeptide) an importance value for a specific prediction. | Identify which amino acid pairs most strongly contribute to a sequence being classified as a specific R-gene class (e.g., CNL vs. TNL). |
| LIME (Local Interpretable Model-agnostic Explanations) [49] | Post-hoc, Model-agnostic | Approximates a complex model locally with an interpretable one (e.g., linear model). | Generate "local" explanations for individual R-gene predictions to understand model reasoning on a case-by-case basis. |
| Inherently Interpretable Models [49] | Model-based | Uses simpler, transparent models by design (e.g., decision trees, linear models). | Serve as a baseline for complex models. Hybrid models that integrate symbolic knowledge (e.g., domain rules) into neural networks are a promising direction. |
| Feature Attribution Visualization [49] | Post-hoc, Model-specific | Creates visual highlights of important input regions (e.g., saliency maps). | Visualize which regions of a protein sequence the model "attends to," potentially highlighting key functional domains. |
Experimental Protocol: Explaining Predictions with SHAP
This protocol provides a step-by-step method for applying SHAP to interpret predictions from a trained R-gene classifier, using the PRGminer model as an example.
Objective: To explain the predictions of a deep learning R-gene classifier by identifying the most influential dipeptide features. Materials: A trained deep learning model for R-gene prediction (e.g., a saved PRGminer model or a custom Keras/PyTorch model); a set of query protein sequences for explanation; the SHAP Python library.
Model and Data Preparation:
- Load your pre-trained model and ensure it is in evaluation mode.
- Preprocess the query sequences identically to the training phase (i.e., convert them to their dipeptide composition representation).
SHAP Explainer Initialization:
- Select an appropriate SHAP explainer. For deep learning models,
DeepExplainerorGradientExplainerare commonly used. - Initialize the explainer by passing it your trained model and a representative background dataset (e.g., a random subset of 100-200 sequences from your training set). This background dataset is used to integrate out the effect of features.
- Select an appropriate SHAP explainer. For deep learning models,
Calculating SHAP Values:
- Compute the SHAP values for the set of query sequences you wish to explain.
- The output will be a matrix of SHAP values, with the same dimensions as your input feature matrix (number of sequences x number of dipeptide features). Each value represents the contribution of a specific dipeptide to the final prediction for that sequence.
Interpretation and Visualization:
- Global Interpretation: To understand what the model has learned overall, create a summary plot of the SHAP values for all query sequences. This plot ranks the dipeptide features by their average impact on the model output.
- Local Interpretation: To explain a single prediction, use a force plot or waterfall plot. This visualization shows how the model's base value (average prediction) was pushed to the final output by the contributions of the most important dipeptides for that specific sequence. Correlate high-impact dipeptides with known protein domains to generate biologically testable hypotheses.
Diagram 1: SHAP Interpretation Workflow
Application Note: Accounting for Pathogen Evolution
Pathogens are not static; they evolve rapidly to overcome plant resistance, often through mutations in their effector proteins that prevent recognition by R-proteins [14]. A major limitation of purely sequence-based prediction models is their inability to account for this evolutionary arms race. Therefore, computational predictions must be validated with strategies that consider pathogen diversity and evolution.
Table 3: Evolutionary-Aware Validation Techniques for Predicted R-genes
| Validation Technique | Description | Information Gained |
|---|---|---|
| Effector Binding Site Prediction | Use computational tools to predict the putative effector-binding interface on the LRR domain of a newly predicted R-gene. | Identifies potential sites under positive selection; residues critical for pathogen recognition. |
| Positive Selection Analysis | Calculate the ratio of non-synonymous to synonymous substitutions (dN/dS) across homologs of the predicted R-gene in a population. | A dN/dS > 1 indicates diversifying selection, a hallmark of genes involved in co-evolutionary arms races. |
| In silico Effector Co-evolution | Analyze if the predicted R-gene shows signatures of co-evolution with effector proteins from a specific pathogen. | Provides circumstantial evidence for a specific gene-for-gene interaction and can narrow down the potential pathogen target. |
Experimental Protocol: In silico Positive Selection Analysis
This protocol describes a method to detect signatures of positive selection in a cluster of R-genes containing a novel predicted gene, providing evolutionary evidence for its functional role.
Objective: To test whether a genomic region containing a predicted R-gene is under positive selection, indicative of an evolutionary arms race with pathogens. Materials: Genome or transcriptome sequences from multiple accessions or related species of the plant of interest; software for sequence alignment (e.g., MAFFT) and positive selection analysis (e.g., CodeML from the PAML package).
Sequence Homolog Identification and Alignment:
- Using the predicted R-gene as a query, perform a BLAST search against the multi-accession sequence database to identify its homologs/orthologs.
- Extract the coding sequences (CDS) of these homologs.
- Perform a multiple sequence alignment of the CDS using a codon-aware aligner, such as that implemented in MAFFT or PRANK.
Phylogenetic Tree Construction:
- Using the aligned CDS, construct a phylogenetic tree using maximum likelihood methods (e.g., RAxML or IQ-TREE). This tree represents the evolutionary relationships among the different homologs.
CodeML Analysis for Site-Specific Selection:
- Prepare the configuration files for CodeML. The analysis will compare two models:
- Null Model (M7): Assumes a beta distribution for dN/dS (ω) ratios between 0 and 1, disallowing positive selection (ω > 1).
- Alternative Model (M8): Allows for a class of sites with ω > 1, which indicates positive selection.
- Run CodeML under both models using the alignment and the phylogenetic tree.
- Prepare the configuration files for CodeML. The analysis will compare two models:
Statistical Testing and Interpretation:
- CodeML will output a log-likelihood value for each model. Perform a Likelihood Ratio Test (LRT) by calculating 2*(lnL(M8) - lnL(M7)), where lnL is the log-likelihood. This test statistic follows a chi-square distribution with degrees of freedom equal to the difference in free parameters between the models (e.g., 2 for M7 vs M8).
- If the LRT is statistically significant (p-value < 0.05), reject the null model and accept that a proportion of sites are under positive selection. The Bayes Empirical Bayes (BEB) analysis under model M8 will identify which specific amino acid sites have a high posterior probability of being under positive selection. Map these sites onto the 3D structure of the R-gene, if available, to see if they cluster in the LRR or other putative effector-binding domains.
Diagram 2: Positive Selection Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Computational Tools and Databases for R-gene Research
| Tool / Database | Type | Function | Relevance to PRGminer Research |
|---|---|---|---|
| PRGminer Webserver [3] [11] | Deep Learning Prediction Tool | High-throughput identification and classification of plant R-genes from protein sequences. | Core tool for initial, rapid screening of proteomes or candidate genes. |
| PRGdb [14] | Curated Database | A centralized resource for known and predicted plant resistance genes. | Source of positive training data and for benchmarking predictions. |
| InterProScan [14] | Domain Annotation Tool | Scans protein sequences against multiple databases to identify functional domains and motifs. | Critical for validating the domain architecture of R-genes predicted by PRGminer. |
| HMMER3 [3] | Domain Search Tool | Uses profile hidden Markov models to identify distant protein homologs and domains. | Used for building custom HMM profiles for specific R-gene classes. |
| SHAP Library [49] | Explainable AI (XAI) Tool | Explains the output of any machine learning model by attributing importance to each input feature. | For interpreting PRGminer predictions and generating biological hypotheses. |
| PAML (CodeML) [14] | Evolutionary Analysis Tool | A package for phylogenetic analysis, including codon-based models of molecular evolution. | For performing positive selection analysis on predicted R-gene clusters. |
| Phytozome [3] | Plant Genomics Resource | Provides access to sequenced and annotated plant genomes and proteomes. | Primary source for retrieving protein sequences for analysis and for negative dataset construction. |
Conclusion
PRGminer represents a significant leap forward in plant genomics, demonstrating how deep learning can overcome the limitations of traditional, homology-based methods for predicting resistance genes. Its high accuracy in both identifying and classifying R-genes into eight distinct functional classes provides researchers with a powerful, scalable tool for deciphering the genetic basis of plant immunity. The successful application of this tool promises to accelerate the pace of R-gene discovery in both model plants and crop species, directly contributing to the development of disease-resistant cultivars. Future advancements will depend on interdisciplinary collaboration to enhance model interpretability, integrate multi-omics data, and expand training datasets. Ultimately, tools like PRGminer are pivotal for building a more resilient global food system through intelligent, data-driven crop improvement strategies.
References
- 1. Plant Immune System: Understanding Pathogen ... [https://premierscience.com/pjs-24-682/]
- 2. Revealing Shared and Distinct Gene Network Organization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4348776/]
- 3. PRGminer: harnessing deep learning for the prediction of ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1411525/full]
- 4. From PTI To ETI: An Overview Of Plant Defence Response [https://global-engage.com/resource-center/from-pti-to-eti-an-overview-of-plant-defence-response/]
- 5. PTI‐ETI synergistic signal mechanisms in plant immunity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11258992/]
- 6. Modular mechanisms of immune priming and growth ... [https://www.sciencedirect.com/science/article/pii/S2211124725001652]
- 7. RNA‐binding proteins orchestrating immunity in plants - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12425495/]
- 8. PRGminer: harnessing deep learning for the prediction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12170542/]
- 9. PRGMiner Tutorial [https://kaabil.net/prgminer/tutorial]
- 10. R gene [https://en.wikipedia.org/wiki/R_gene]
- 11. PRGminer [https://kaabil.net/prgminer/]
- 12. Pathogen perception and signaling in plant immunity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11062475/]
- 13. R Gene - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/r-gene]
- 14. Computational Strategies for Elucidating Plant Disease ... [https://www.sciencedirect.com/science/article/abs/pii/S0885576525003790]
- 15. Concerted expansion and contraction of immune receptor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9579050/]
- 16. A Gain-of-Function Mutation in a Plant Disease Resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC280567/]
- 17. Structure, biochemical function, and signaling mechanism ... [https://www.sciencedirect.com/science/article/pii/S1674205222004117]
- 18. Multi-omics approach highlights differences between RLP ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-021-07855-0]
- 19. DaapNLRSeek, prediction, and evolution of resistance ... [https://www.sciencedirect.com/science/article/pii/S2589004225011307]
- 20. Current Status and Challenges in Identifying Disease ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2017.01788/full]
- 21. Resistance gene identification from Larimichthys crocea ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5138596/]
- 22. comparing deep learning and GBLUP across diverse plant ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1568705/full]
- 23. Integrating AI in plant science: A review of applications and ... [https://www.sciencedirect.com/science/article/abs/pii/S2352407325000538]
- 24. : harnessing PRGminer for the prediction of deep ... learning resistance [https://pubmed.ncbi.nlm.nih.gov/40530297/]
- 25. Artificial Intelligence-Assisted Breeding for Plant Disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12154274/]
- 26. MtCro: multi-task deep learning framework improves multi-trait ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-024-01321-0]
- 27. Machine Learning -Based identification of resistance ... genes [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-025-01383-8]
- 28. Predicting antifreeze proteins with weighted generalized ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04251-z]
- 29. Defended to the Nines: 25 Years of Resistance Gene Cloning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5868693/]
- 30. PRGdb 3.0: a comprehensive platform for prediction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5753367/]
- 31. an R package for visualization the element on bio-sequences [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1645004/full]
- 32. Top 10 Best Practices for Managing and Cleansing Large ... [https://datagroomr.com/top-10-best-practices-for-managing-and-cleansing-large-datasets/]
- 33. Machine Learning Best Practices for Big Dataset [https://datascience.stackexchange.com/questions/13901/machine-learning-best-practices-for-big-dataset]
- 34. Best Public Datasets for Machine Learning in 2025 [https://365datascience.com/trending/public-datasets-machine-learning/]
- 35. 5 Best Machine Learning Repository Datasets (2025) [https://averroes.ai/blog/machine-learning-repository-datasets]
- 36. AI-Powered Plant Immunity Against Bacterial Diseases [https://www.biotechniques.com/microbiology/ai-powered-plant-immunity-could-protect-crops-from-bacterial-diseases/]
- 37. Molecular breakthroughs in modern plant breeding ... [https://www.sciencedirect.com/science/article/pii/S2468014124000311]
- 38. QBMS 2.0: Advancing Data Integration for Modern Plant ... [https://icarda.org/media/news/qbms-20-advancing-data-integration-modern-plant-breeding]
- 39. Prediction of Plant Resistance Proteins Using Alignment ... [https://pubmed.ncbi.nlm.nih.gov/39580673/]
- 40. Machine learning in biological research: key algorithms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574268/]
- 41. AI vs. Machine Learning vs. Deep Learning vs. Neural ... [https://www.ibm.com/think/topics/ai-vs-machine-learning-vs-deep-learning-vs-neural-networks]
- 42. Comparison of machine learning and deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7959599/]
- 43. Deep Learning vs Machine Learning - Difference Between ... [https://aws.amazon.com/compare/the-difference-between-machine-learning-and-deep-learning/]
- 44. Deep Learning vs. Traditional Machine Learning: Which is ... [https://www.alliancetek.com/blog/post/2025/03/11/deep-learning-vs-traditional-ml.aspx]
- 45. Deep Learning vs Machine Learning: The Ultimate Battle. [https://www.turing.com/kb/ultimate-battle-between-deep-learning-and-machine-learning]
- 46. R we there yet? Advances in cloning resistance genes for ... [https://www.sciencedirect.com/science/article/pii/S1369526623001541]
- 47. Genome wide association study reveals new genes for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11427250/]
- 48. Genome-wide association study reveals the genetic basis ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2024.1476829/full]
- 49. Investigating the Duality of Interpretability and ... [https://arxiv.org/html/2503.21356v1]
- 50. Interpretable machine learning methods for predictions in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9650551/]