Genomic vs. Transcriptomic Prediction Models: A Comprehensive Performance Comparison for Biomedical Research
This article provides a comprehensive performance comparison of genomic and transcriptomic prediction models, tailored for researchers, scientists, and drug development professionals.
Genomic vs. Transcriptomic Prediction Models: A Comprehensive Performance Comparison for Biomedical Research
Abstract
This article provides a comprehensive performance comparison of genomic and transcriptomic prediction models, tailored for researchers, scientists, and drug development professionals. It explores the foundational principles of both genomic selection and transcriptomic data, detailing how they capture different layers of biological information. The content covers a wide array of methodological approaches, from traditional BLUP models to advanced multi-omics integration and machine learning techniques. It addresses key challenges in model implementation, including data redundancy and technical complexity, while offering optimization strategies. Through rigorous validation and comparative analysis across diverse applications—from agriculture to drug response prediction and personalized medicine—this article synthesizes evidence on the complementary strengths of each approach and provides actionable insights for selecting and refining predictive models in research and development.
Core Concepts: Understanding Genomic and Transcriptomic Data for Predictive Modeling
Genomic Selection (GS) has revolutionized animal and plant breeding by enabling the prediction of an individual's genetic merit based on genome-wide molecular markers. First proposed by Meuwissen et al. in 2001, GS bypasses the need for direct phenotypic selection, allowing for earlier and more efficient selection decisions that shorten breeding cycles and enhance genetic gain [1] [2]. This methodology represents a fundamental shift from phenotype-based to genotype-driven decision-making in breeding programs. The core principle involves developing a prediction model using genotypic and phenotypic data from a training population, which is then applied to estimate Genomic Estimated Breeding Values (GEBVs) for individuals in a breeding population based solely on their genomic profiles [2].
In recent years, attention has turned toward other omics layers seen as promising for improving prediction accuracy. Transcriptomic data, which provides insights into gene expression patterns shaped by both genetic and environmental factors, offers a more comprehensive understanding of phenotype expression [3]. This article provides a comprehensive comparison between traditional genomic prediction and emerging transcriptomic prediction approaches, examining their relative performances across species, traits, and experimental conditions to guide researchers in selecting appropriate strategies for genetic improvement.
Performance Comparison: Genomic vs. Transcriptomic Prediction
Direct comparisons between genomic and transcriptomic prediction models across multiple studies reveal a complex performance landscape influenced by species, trait characteristics, and environmental conditions. The table below summarizes key findings from recent research:
Table 1: Comparison of Genomic and Transcriptomic Prediction Accuracies Across Studies
| Species | Traits Assessed | Genomic Prediction Accuracy | Transcriptomic Prediction Accuracy | Combined Model Accuracy | Reference |
|---|---|---|---|---|---|
| Japanese Quail | Efficiency-related traits (P utilization, body weight) | Moderate | Higher than genomic | Highest | [3] |
| Wheat | Flowering time, height | Moderate | Superior in controlled environments | Best performing | [4] |
| Barley | Agricultural traits | 0.73-0.78 (50K SNP array) | Comparable to genomic | 0.73-0.78 (consensus SNP) | [5] |
| Dairy Cattle | Lactation traits | Baseline | Functional variants from RNA-seq improved accuracy | Varied by trait | [6] |
| Maize & Rice | Complex agronomic traits | Variable | Complementary to genomic | Consistently improved | [1] |
Transcriptomic data generally explains a larger portion of phenotypic variance than host genetics for many traits. In Japanese quail, transcript abundances from intestinal tissue explained more phenotypic variance of efficiency-related traits than genetic markers [3]. Similarly, in wheat grown under controlled environments, transcriptome abundance outperformed genomic data when considered independently for predicting flowering time and height [4].
However, the superior predictive ability of transcriptomic data is context-dependent. In field conditions with greater environmental variability, the relative advantage of transcriptomic data diminishes while models combining genomic and environmental data often provide comparable gains at lower cost [4]. For some traits, particularly those with well-characterized genetic architecture, genomic data may remain superior, as seen with yield traits in maize where genomic data outperformed transcriptomic and metabolomic layers [4].
Methodological Approaches and Experimental Protocols
Statistical Models for Prediction
Various statistical approaches have been employed for genomic and transcriptomic prediction:
- GBLUP (Genomic Best Linear Unbiased Prediction): Uses a genomic relationship matrix derived from SNP markers to capture additive genetic effects [3] [4]
- TBLUP (Transcriptomic BLUP): Applies the BLUP framework to transcript abundance data to predict phenotypes [3]
- GTBLUP: Incorporates both genomic and transcriptomic data as independent random effects [3]
- GTCBLUP/GTCBLUPi: Advanced models that account for redundant information between genomic and transcriptomic data by conditioning transcriptomic effects on genetics [3]
- RKHS (Reproducing Kernel Hilbert Spaces): A semi-parametric method using Gaussian kernel functions to capture non-linear relationships [7] [4]
- Bayesian Models (e.g., BayesA, BayesB, BayesCπ): Allow for different prior distributions of marker effects [7] [6]
- Machine Learning Approaches: Including random forests, gradient boosting, and deep learning architectures [1] [7]
Experimental Workflows
Standardized experimental protocols have emerged for comparative studies:
Table 2: Key Methodological Components in Prediction Studies
| Component | Genomic Prediction Approach | Transcriptomic Prediction Approach |
|---|---|---|
| Data Generation | SNP arrays, GBS, WGS | RNA-Seq, microarrays, Fluidigm BioMark |
| Data Processing | Quality control, imputation, MAF filtering | Normalization, quality control, transformation |
| Model Training | Training population with genotypes and phenotypes | Training population with transcriptomes and phenotypes |
| Validation | Cross-validation, independent validation sets | Cross-validation, independent validation sets |
| Assessment | Correlation between predicted and observed phenotypes | Correlation between predicted and observed phenotypes |
The typical workflow begins with careful experimental design. For transcriptomic studies, this includes standardized cultivation conditions, precise timing of tissue collection, and high-throughput RNA extraction methods [5]. In barley research, researchers cultivated all recombinant inbred lines under controlled conditions in vertically stacked square Petri dishes for seven days in reach-in growth chambers with fixed temperature, humidity, and light intensity [5]. RNA extraction typically uses TRIzol reagent with adaptations for 96-well formats to enable high-throughput processing [5].
Library preparation for RNA-Seq has been miniaturized to reduce costs, with studies successfully reducing reagent volumes to 25% of original amounts without compromising data quality [5]. For genomic studies, DNA extraction followed by genotyping using platforms such as Illumina SNP chips or genotyping-by-sequencing represents the standard approach.
Figure 1: Experimental workflow for genomic and transcriptomic prediction studies
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful implementation of genomic and transcriptomic prediction requires specific research reagents and platforms:
Table 3: Essential Research Reagents and Platforms for Prediction Studies
| Category | Specific Tools/Reagents | Function | Example Applications |
|---|---|---|---|
| Genotyping Platforms | Illumina SNP chips, Genotyping-by-Sequencing | Genome-wide marker identification | Genetic relationship matrix construction [5] [2] |
| Transcriptomics Technologies | RNA-Seq, Fluidigm BioMark HD system | Gene expression quantification | Transcript abundance measurement [3] [5] |
| Library Preparation Kits | VAHTS Universal V6 RNA-seq Library Prep Kit | cDNA library construction | Preparation for sequencing [5] |
| RNA Extraction Reagents | TRIzol reagent | High-quality RNA isolation | Tissue RNA extraction [5] |
| Sequencing Platforms | Illumina systems | High-throughput sequencing | Genotype and expression data generation [5] |
| Analysis Software | ASReml R, JWAS, EasyGeSe | Statistical modeling and prediction | Implementation of BLUP and Bayesian models [3] [7] [6] |
The Fluidigm BioMark HD system has been particularly valuable for high-throughput transcriptomic studies, enabling efficient quantification of candidate transcripts across hundreds of individuals [3]. For RNA extraction, TRIzol reagent with adaptations for 96-well formats allows processing of large sample sizes essential for robust prediction modeling [5].
Recent advances in benchmarking tools such as EasyGeSe provide standardized datasets and evaluation procedures for comparing prediction methods across diverse species [7]. This resource encompasses data from multiple species including barley, maize, rice, and wheat, enabling more reproducible comparisons of genomic prediction methods [7].
Integration Strategies and Multi-Omics Approaches
The integration of genomic and transcriptomic data often outperforms models using either data type alone. Several integration strategies have been developed:
- Early Fusion (Data Concatenation): Combining genomic and transcriptomic data before model construction [1]
- Model-Based Integration: Advanced frameworks that capture non-additive, nonlinear, and hierarchical interactions across omics layers [1]
- Conditioned Models (GTCBLUPi): Models that explicitly account for redundancy between genomic and transcriptomic information [3]
Research comparing 24 integration strategies combining genomics, transcriptomics, and metabolomics found that model-based fusion methods consistently improved predictive accuracy over genomic-only models, particularly for complex traits [1]. In contrast, several commonly used concatenation approaches did not yield consistent benefits and sometimes underperformed [1].
The GTCBLUPi model, which addresses redundant information between genomic and transcriptomic data, has proven to be a suitable framework for integration [3]. This approach conditions transcriptomic effects on genetic effects, ensuring that the transcriptomic components captured are purely non-genetic, thereby avoiding collinearity problems [3].
Figure 2: Multi-omics integration strategies for enhanced prediction accuracy
Genomic selection using genome-wide markers has established itself as a powerful tool for predicting complex traits in plant and animal breeding. The integration of transcriptomic data provides complementary information that often enhances prediction accuracy, particularly for traits influenced by gene regulation and environmental responses. While transcriptomic data alone frequently explains more phenotypic variance than genomic data, the most effective strategies combine both data types using advanced integration methods that account for their redundant information.
The choice between genomic and transcriptomic approaches depends on multiple factors including trait architecture, environmental influence, resource availability, and breeding objectives. For commercial breeding programs, the higher costs and complexity of generating transcriptomic data may currently limit its feasibility, though combining genomic data with well-characterized environmental covariates provides a practical alternative with similar gains. As sequencing costs continue to decline and multi-omics integration methods improve, the combined use of genomic and transcriptomic information holds significant promise for accelerating genetic improvement across agricultural species.
The transition from static genetic blueprints to dynamic, observable traits represents one of the most significant challenges in modern biology. While genomic data provides a comprehensive catalog of inherited variants, it offers limited insight into how these variants dynamically orchestrate molecular processes that ultimately manifest as phenotypes. Transcriptomic data, which captures the full complement of RNA molecules in a cell, serves as a crucial functional intermediary that bridges this fundamental gap between genotype and phenotype [8]. This comparative analysis examines the relative performance of genomic and transcriptomic prediction models across multiple biological contexts, demonstrating how transcriptomics provides a more direct, functional readout of cellular states that enhances phenotypic prediction accuracy.
The limitations of single-omics approaches have become increasingly apparent in complex trait prediction. Genomics alone cannot quantify the spatiotemporal specificity of gene expression or its regulatory mechanisms [8]. Furthermore, genetic variants often exert their effects through subtle changes in gene regulation rather than through direct protein-coding changes. Transcriptomics addresses these limitations by capturing the integrated effects of genetic variation, environmental influences, and regulatory mechanisms, providing a more comprehensive understanding of the molecular networks underlying phenotypic diversity [8] [9].
Theoretical Foundations: Molecular Hierarchies from Genome to Phenome
The Central Dogma and Transcriptomic Positioning
The flow of genetic information follows a fundamental pathway from DNA to RNA to protein, with transcriptomics occupying the critical intermediate position in this biological cascade. While DNA represents the static code, the transcriptome reflects dynamically regulated processes including transcription, RNA processing, and degradation that collectively determine functional outputs [8]. This positioning enables transcriptomic data to capture both genetic influences and environmental perturbations that collectively shape phenotypic outcomes.
Transcriptomic profiling moves beyond mere sequence information to reveal how genes are quantitatively regulated across different conditions, tissues, and timepoints. This regulatory dimension provides critical functional context that static DNA sequences lack. As noted in epilepsy research, "genomics identify candidate disease-causing genes for epilepsy, but it cannot quantify their expression levels" [8]. The integration of transcriptomics elucidates the spatiotemporal specificity of gene expression and its regulatory mechanisms, providing a more complete picture of molecular networks underlying different epilepsy phenotypes.
Transcriptional Regulation of Complex Traits
The relationship between transcript abundance and phenotypic outcomes is governed by complex regulatory networks involving transcription factors, non-coding RNAs, and epigenetic modifications. Studies in mango fruit development demonstrated how transcription factors MibZIP66 and MibHLH45 activate MiPSY1 transcription by directly binding to the CACGTG motif of the MiPSY1 promoter, thereby regulating β-carotene biosynthesis and affecting fruit flesh color [10]. Such mechanistic insights are only possible through integrated analysis that includes transcriptomic data.
The transcriptome's responsiveness to both internal genetic programs and external environmental cues makes it particularly valuable for predicting dynamic traits. In quail research, transcript abundances from the ileum explained a larger portion of the phenotypic variance for efficiency-related traits than host genetics alone [3]. This demonstrates how transcriptomics captures the functional integration of multiple influences that collectively determine phenotypic outcomes.
Experimental Comparison: Genomic vs. Transcriptomic Prediction Models
Direct Performance Comparison in Avian Models
A comprehensive study in Japanese quail (Coturnix japonica) provides compelling direct evidence comparing genomic and transcriptomic prediction models for efficiency-related traits [3]. Researchers utilized various statistical methods including GBLUP (genomic best linear unbiased prediction), TBLUP (transcriptomic BLUP), and integrated models to predict phenotypes including phosphorus utilization (PU), body weight gain (BWG), feed intake (FI), feed conversion ratio (FCR), tibia ash amount (TA), and calcium utilization (CaU).
Table 1: Prediction Accuracy Comparison of Genomic and Transcriptomic Models for Quail Efficiency Traits
| Trait | GBLUP (Genomic Only) | TBLUP (Transcriptomic Only) | GTBLUP (Combined) |
|---|---|---|---|
| Phosphorus Utilization (PU) | Lower accuracy | Higher accuracy | Highest accuracy |
| Body Weight Gain (BWG) | Lower accuracy | Higher accuracy | Highest accuracy |
| Feed Intake (FI) | Lower accuracy | Higher accuracy | Highest accuracy |
| Feed Conversion Ratio (FCR) | Lower accuracy | Higher accuracy | Highest accuracy |
| Tibia Ash (TA) | Lower accuracy | Higher accuracy | Highest accuracy |
| Calcium Utilization (CaU) | Lower accuracy | Higher accuracy | Highest accuracy |
The study demonstrated that "transcript abundances from the ileum explain a larger portion of the phenotypic variance of the traits than host genetics" across all measured efficiency traits [3]. Importantly, models incorporating both genetic and transcriptomic information (GTBLUP) consistently outperformed models using either data type alone, confirming that transcriptomic information complements genetic data effectively rather than simply replicating it.
Methodological Framework for Model Comparison
The experimental protocol for direct model comparison followed rigorous statistical standards [3]:
Population Design: 480 F2 cross Japanese quail selected from an initial total of 920 animals, raised under controlled conditions with standardized diet during the strong growing phase between days 10-15 of life.
Phenotyping: Comprehensive efficiency measurements including PU based on total P intake and P excretion, BWG between days 10-15, FI during the 5-day period, FCR as FI divided by BWG, TA in mg, and CaU based on total Ca intake and Ca excretion.
Genotyping: 4k SNPs after filtering using a 6k Illumina iSelect chip with established genetic linkage map.
Transcriptomic Profiling: Ileal miRNA and mRNA sequencing followed by candidate assessment with 96.96 dynamic arrays on a Fluidigm BioMark HD system.
Statistical Analysis: Box-Cox transformation of phenotypic data with trait-specific lambda parameters followed by BLUP model comparisons including:
- GBLUP: y = Xb + Zgg + e
- TBLUP: y = Xb + Ztt + e
- GTBLUP: y = Xb + Zgg + Ztt + e
- GTCBLUP: Integrated model addressing redundancy between genomic and transcriptomic information
The mathematical framework for the integrated GTCBLUP model was specifically derived to handle the overlapping nature of genomic and transcriptomic data layers, preventing collinearity problems that would arise from treating them as independent random effects [3].
Diagram 1: Transcriptomic data bridges DNA and phenotype, capturing dynamic functional information that static genetic data misses. The bold pathway highlights transcriptomics' direct predictive power for phenotypic outcomes.
Case Studies Across Biological Systems
Neurological Disorders: Epilepsy Research Applications
In epilepsy research, multi-omics approaches have revealed the complex molecular dysregulation networks underlying different epilepsy phenotypes [8]. The transition from traditional hypothesis-driven research to data-driven architectures has been catalyzed by multi-omics methods, with transcriptomics playing a crucial role in understanding the functional consequences of genetic variants associated with epilepsy susceptibility.
Despite the availability of over 20 anti-seizure medications, about one-third of epilepsy patients develop drug-resistant epilepsy [8]. Transcriptomic profiling has helped identify molecular subtypes that may explain this treatment resistance, moving beyond the limitations of purely genetic classification. The integrated analysis of transcriptomic data with genomic findings has provided insights into the spatiotemporal specificity of gene expression and its regulatory mechanisms in neurological tissues.
Agricultural Genomics: Crop Improvement Applications
In mango fruit research, chromosome-scale genome assembly combined with comparative transcriptomic analysis identified transcriptional regulators of β-carotene biosynthesis [10]. Researchers compared β-carotene content in two different cultivars ("Irwin" and "Baixiangya") across growth periods, finding that variation in β-carotene content mainly affected fruit flesh color.
Transcriptome analysis identified MiPSY1 as a key gene regulating β-carotene biosynthesis, with subsequent functional validation confirming that transcription factors MibZIP66 and MibHLH44 activate MiPSY1 transcription by directly binding to the CACGTG motif of the MiPSY1 promoter [10]. This mechanistic understanding of fruit quality traits demonstrates how transcriptomics bridges the gap between genomic sequences and commercially relevant phenotypic traits.
Parasitology: Helminth Genome Biology
In Haemonchus contortus research, genomic and transcriptomic variation analysis defined the chromosome-scale assembly of this model gastrointestinal worm [11]. The integration of transcriptomic data allowed researchers to define coordinated transcriptional regulation throughout the parasite's life cycle and refine understanding of cis- and trans-splicing.
The remarkable pattern of chromosome content conservation with Caenorhabditis elegans, despite almost no conservation of gene order, highlights the importance of transcriptomic data for understanding functional genomics in parasitic species [11]. This comparative approach provides insights into evolutionarily conserved operons and regulatory mechanisms that would be inaccessible through genomic analysis alone.
Technical Considerations in Transcriptomic Experimentation
Methodological Best Practices and Pitfalls
Transcriptomic experimentation requires careful consideration of multiple technical factors to ensure data quality and biological relevance [12]:
Experimental Design: Statistical countermeasures must be implemented throughout experimentation, including proper randomization, sufficient replicates, and appropriate statistical methods such as false discovery rate correction. Inadequate implementation due to budget constraints or lack of statistical expertise frequently undermines experimental outcomes.
Sample Pooling Decisions: While pooling samples intuitively seems to average out differences between individuals, it actually eliminates the variation needed for statistical power and inference. Pooling substantially different cells creates artificial in-between cell types that can hamper biological interpretation.
Perturbation Severity: Severe perturbations often trigger generic stress responses that obscure specific reactions to the perturbation of interest. Range-finding experiments help determine optimal experimental settings that elicit specific responses without overwhelming generic stress pathways.
Technical vs Biological Replication: Biological variation heavily outweighs technological variation in transcriptomics, making biological replicates generally more valuable than technical replicates despite lingering preferences from early microarray technology.
Analytical Frameworks and Visualization Approaches
Effective visualization of transcriptomic data is essential for exploring large datasets and uncovering hidden patterns [13]. Different visualization approaches serve distinct analytical purposes:
Table 2: Transcriptomic Data Visualization Methods and Applications
| Visualization Method | Data Type | Primary Application | Strengths |
|---|---|---|---|
| Volcano Plot | Differential expression | Significance vs magnitude of change | Identifies statistically significant large-effect changes |
| Heatmap | Gene expression matrix | Multi-sample expression patterns | Visualizes expression patterns across many samples/genes |
| Violin Plot | Single-cell expression | Distribution of expression values | Shows full distribution rather than summary statistics |
| Network Visualization | Gene interactions | Regulatory relationships | Maps complex interaction networks between genes |
| Pathway Diagrams | Enrichment results | Biological process visualization | Contextualizes results within known biological pathways |
Space-filling layouts such as Hilbert curves preserve the sequential nature of genomic features while allowing visual integration of multiple datasets [13]. Circular layouts like Circos plots efficiently display sequences and interactions in a space-saving manner, enabling simultaneous visualization of multiple data types including mutations, copy number changes, and translocations.
Research Reagent Solutions for Transcriptomic Studies
Table 3: Essential Research Reagents for Transcriptomic Experiments
| Reagent/Category | Specific Examples | Function | Application Context |
|---|---|---|---|
| Sequencing Platforms | Illumina NovaSeq X, Oxford Nanopore | High-throughput sequencing | Whole transcriptome sequencing, targeted RNA-seq |
| Single-cell RNA-seq | 10X Genomics, Fluidigm | Cellular resolution transcriptomics | Tumor heterogeneity, developmental biology |
| Spatial Transcriptomics | 10X Genomics Visium | Tissue context preservation | Brain mapping, tumor microenvironment |
| qPCR Validation | Fluidigm BioMark HD | Targeted expression validation | Candidate gene verification, biomarker confirmation |
| Library Preparation | Illumina, Takara Bio | RNA library construction | Strand-specific RNA-seq, small RNA sequencing |
The research utilized specific reagent systems including 96.96 dynamic arrays on a Fluidigm BioMark HD system for assessing miRNA and mRNA candidates [3]. For single-cell and spatial transcriptomics, the 10X Genomics Visium platform has been commercialized and widely adopted for preserving spatial context in transcriptomic measurements [8].
Integrated Multi-Omics Prediction Frameworks
Statistical Models for Data Integration
The redundancy between different molecular data layers presents statistical challenges for integrated models. The GTCBLUP model addresses this by conditioning transcriptomic effects on genetic effects to remove shared variation [3]. This approach models genotype data and omics data conditioned on the genotypes simultaneously in a one-step approach, ensuring that the modeled omics effects are purely non-genetic.
Alternative approaches include the two-step procedure proposed by Christensen et al. that first estimates the total effect of omics data on phenotypes and then explicitly models the genetic portion of these omics effects in a second step [3]. The optimal approach depends on the specific research question and the nature of the genetic control over transcriptomic features.
Functional Genomics in Psychiatric Research
Novel approaches in psychiatric research have begun incorporating functional molecular phenotypes that are closer to genetic variation and less penalized by multiple testing burdens [9]. Moving from genotype-disease to genotype–gene regulation frameworks, these approaches incorporate prior knowledge regarding biological processes involved in disease and aggregate estimates for the association of genotypes and phenotypes using multi-omics data modalities.
This shift from traditional polygenic risk scores to functionally informed risk assessment demonstrates how transcriptomic data provides biological context for genetic signals, helping generate biologically driven hypotheses that can ultimately serve as potential biomarkers of disease susceptibility [9].
Diagram 2: Multi-omics integration framework with transcriptomic data as a central component. Statistical models like GTCBLUP handle redundancy between data layers to enhance phenotypic prediction accuracy.
The comparative analysis of genomic and transcriptomic prediction models demonstrates the superior performance of transcriptomic data for predicting complex phenotypes across multiple biological systems. Transcriptomics serves as a functional intermediary that captures the dynamic integration of genetic predispositions, environmental influences, and regulatory mechanisms that collectively determine phenotypic outcomes.
While transcriptomic data alone explains a larger portion of phenotypic variance than genetic data alone, the most accurate predictions come from integrated models that leverage both data types [3]. The complementary nature of genomic and transcriptomic information reflects the biological reality that DNA sequence provides the template, while RNA expression reflects the functional implementation of that template in specific contexts.
Future directions in transcriptomic prediction will likely include greater incorporation of single-cell and spatial resolution data, longitudinal profiling to capture dynamic processes, and integration with emerging omics layers including proteomics and metabolomics [8] [14]. As analytical methods continue to evolve, transcriptomic data will remain a cornerstone of predictive biology, providing the critical functional link between genetic inheritance and observable traits across basic research, clinical applications, and agricultural improvement.
In the field of modern genetics, two powerful paradigms offer distinct yet complementary insights into the complex journey from genotype to phenotype. The static genetic blueprint, explored through genomics, provides a comprehensive map of an organism's entire DNA sequence, including its genes and regulatory regions. This blueprint is largely fixed throughout an organism's lifetime. In contrast, dynamic expression profiles, studied via transcriptomics, capture the ever-changing set of RNA transcripts present in a cell at a specific moment, reflecting real-time gene activity in response to developmental cues, environmental stimuli, and disease states [15].
The primary distinction lies in their fundamental nature: genomics offers a static inventory of potential, while transcriptomics reveals the dynamic execution of that potential [15]. For researchers and drug development professionals, understanding the comparative strengths of these approaches is crucial for selecting the appropriate methodology for specific applications, from predicting complex traits in breeding programs to unraveling disease mechanisms for therapeutic discovery. This guide objectively compares their performance, supported by experimental data and detailed methodologies.
Fundamental Characteristics and Comparative Strengths
The table below summarizes the core characteristics that differentiate genomics and transcriptomics.
Table 1: Fundamental Characteristics of Genetic Blueprints and Expression Profiles
| Feature | Static Genetic Blueprint (Genomics) | Dynamic Expression Profiles (Transcriptomics) |
|---|---|---|
| Definition | Study of the complete set of DNA (genome) in an organism [15] | Study of the complete set of RNA transcripts (transcriptome) in a cell at a given time [15] |
| Primary Focus | Genetic structure, sequence, variation, and coding potential [15] | Gene expression levels, activity, and regulation [15] |
| Temporal Nature | Largely static and constant throughout life [15] | Highly dynamic, changing rapidly in response to conditions [15] |
| Key Data Type | DNA sequence, single nucleotide polymorphisms (SNPs), structural variants | RNA sequence counts (mRNA, non-coding RNA), expression levels |
| Information Provided | Genetic blueprint and predisposition | Functional, real-time view of cellular state and response |
Performance Comparison in Predictive Modeling
Genomic Selection (GS), which uses genome-wide markers to predict breeding values, has revolutionized plant and animal breeding [16] [17]. However, its accuracy can be limited for complex traits influenced by regulation and environment. Integrating transcriptomic data aims to capture this missing information, bridging the gap between DNA and phenotype [16] [17].
Recent studies have directly compared the predictive power of genomic and transcriptomic data. The following table summarizes key findings from experiments in animal and plant models.
Table 2: Experimental Comparison of Prediction Accuracy for Complex Traits
| Study Model | Trait Category | Prediction Model | Key Finding on Predictive Power |
|---|---|---|---|
| Japanese Quail [16] | Efficiency traits (e.g., Phosphorus Utilization, Feed Conversion Ratio) | GBLUP (Genomic) | Genomic data explained a portion of the phenotypic variance. |
| TBLUP (Transcriptomic) | Transcript abundances explained a larger portion of the phenotypic variance than host genetics. | ||
| GTCBLUPi (Combined) | Models combining both data types outperformed those using only one type of information. | ||
| Barley RIL Population [5] | Agronomic traits (8 traits across up to 7 environments) | SNP Array (Genomic) | Served as a benchmark for prediction ability. |
| RNA-Seq SNP Data (Transcriptomic) | Achieved prediction ability comparable to or better than the traditional SNP array. | ||
| Consensus SNP (RNA-Seq + WGS) | Performed best, with significant improvements for 5 out of 8 traits and in inter-population predictions. |
Interpretation of Experimental Data
The data consistently demonstrates that transcriptomic information accounts for a significant and often greater portion of phenotypic variance compared to static genomic markers alone [16] [5]. This is because gene expression is shaped by both genetic makeup and environmental factors, providing a more comprehensive view of the biological processes leading to the final phenotype [16].
Furthermore, the most accurate predictions are consistently achieved by models that integrate both genomic and transcriptomic data [16] [17]. This synergy occurs because the two data layers capture complementary information: the static blueprint provides the underlying genetic potential, while the dynamic profile reveals how that potential is being executed in a specific context.
Detailed Experimental Protocols
To ensure reproducibility and a deep understanding of the compared data, here are the detailed methodologies from the key studies cited.
Protocol: Genomic and Transcriptomic Prediction in Japanese Quail
This protocol is adapted from the study that developed the GTCBLUPi model [16].
- Experimental Population and Phenotyping: Create an F2 cross population (e.g., 480 Japanese quails). Raise animals under controlled conditions. Record efficiency-related phenotypes such as Phosphorus Utilization (PU), Body Weight Gain (BWG), and Feed Conversion Ratio (FCR) during a strong growing phase [16].
- Genotyping: Collect blood samples. Genotype all animals using a platform like a 6k Illumina iSelect chip. Filter SNPs for quality, resulting in a final set of several thousand markers [16].
- Transcriptome Sampling: On a predetermined day (e.g., day 15), sacrifice animals and collect tissue samples of interest (e.g., ileum mucosa). Immediately preserve samples for RNA extraction [16].
- RNA Sequencing and Candidate Selection: Extract total RNA. Perform miRNA and mRNA sequencing. Identify differentially expressed transcripts between animals with high and low phenotypes to create a set of candidate miRNAs and mRNAs [16].
- High-Throughput Transcript Quantification: Quantify the selected candidate transcripts across all individuals in the study population using a high-throughput system like a Fluidigm BioMark HD with dynamic arrays [16].
- Data Transformation: Apply a Box-Cox transformation to phenotypic data to normalize distributions and stabilize variance [16].
- Statistical Modeling and Prediction: Construct and compare multiple prediction models using mixed linear models in a software environment like ASReml-R:
- GBLUP: Uses the genomic relationship matrix (G) derived from SNP data.
- TBLUP: Uses a transcriptomic relationship matrix derived from miRNA or mRNA abundance data.
- GTCBLUPi: An integrated model that uses both genomic and transcriptomic matrices, explicitly accounting for the redundancy between them to isolate the non-genetic transcriptomic effects [16].
- Model Validation: Compare models based on the proportion of phenotypic variance explained and the accuracy of predicting phenotypes in validation sets [16].
Protocol: Transcriptome-Based Prediction in Barley
This protocol is adapted from the study on a barley multi-parent RIL population [5].
- Plant Cultivation: Cultivate Recombinant Inbred Lines (RILs) in a randomized, controlled environment. Use standardized conditions for light, temperature, and humidity. Harvest whole seedlings at a specific developmental stage (e.g., 7 days) as a bulk sample for each line [5].
- High-Throughput RNA Extraction: Freeze and grind plant material. Perform total RNA extraction using a miniaturized, high-throughput protocol (e.g., a 96-well format with reduced reagent volumes like a TRIzol-based method) [5].
- Library Preparation and Sequencing: Construct mRNA sequencing libraries using a poly-A tail capture method and a kit like the VAHTS Universal V6 RNA-seq Library Prep Kit. Miniaturize the library preparation process to reduce costs. Sequence the libraries on an Illumina platform [5].
- Data Processing and Genotype Calling:
- Gene Expression Matrix: Map RNA-Seq reads to a reference genome and quantify read counts per gene to create a gene expression matrix.
- RNA-Seq SNP Dataset: Call sequence variants (SNPs) from the RNA-Seq data itself.
- Consensus SNP Dataset: Integrate the RNA-Seq SNPs with a high-quality Whole-Genome Sequencing (WGS) dataset from the parents to create a refined, consensus SNP set [5].
- Phenotypic Data Collection: Measure agronomic traits (e.g., yield components, disease resistance) across multiple field environments to obtain robust phenotypic values [5].
- Genomic Prediction Modeling: Use the different data types (Gene Expression, RNA-Seq SNPs, Consensus SNPs) to build genomic prediction models. A standard benchmark is comparison against predictions from a traditional SNP array [5].
- Model Evaluation: Use cross-validation (e.g., fivefold) to evaluate prediction ability (correlation between predicted and observed phenotypes). Assess both within-population and inter-population prediction scenarios [5].
Visualizing the Workflows
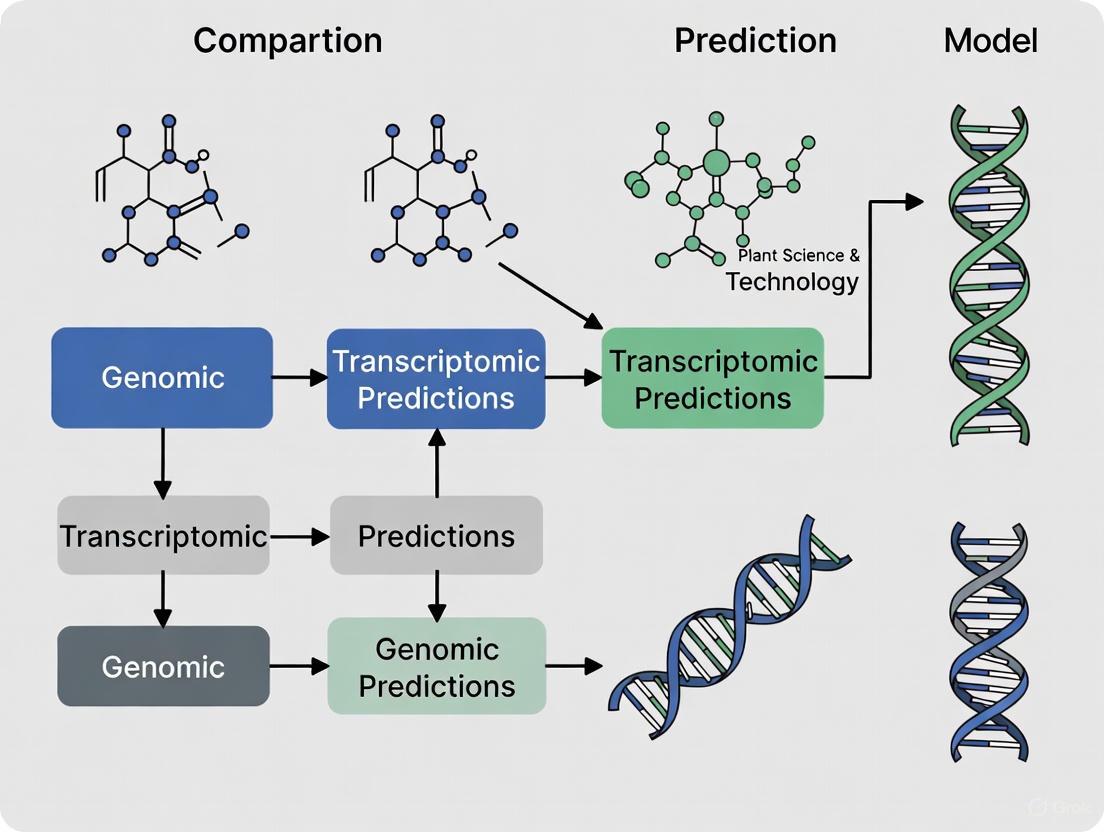
The following diagram illustrates the core workflows for generating and using static genetic blueprints and dynamic expression profiles in predictive modeling, highlighting their convergence in multi-omics integration.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below lists key reagents and materials essential for conducting experiments in genomics and transcriptomics, as derived from the cited protocols.
Table 3: Essential Research Reagents and Solutions for Genomic and Transcriptomic Studies
| Item Name | Function/Application | Example Use Case |
|---|---|---|
| Illumina iSelect Chip | A genotyping array for high-throughput genome-wide SNP profiling. [16] | Genotyping Japanese quail for genomic prediction models (GBLUP). [16] |
| TRIzol Reagent | A ready-to-use monophasic solution for the isolation of high-quality total RNA from cells and tissues. [5] | High-throughput RNA extraction from barley seedling tissue. [5] |
| Fluidigm BioMark HD System | A high-throughput microfluidic platform for targeted gene expression analysis using nano-scale quantitative PCR. [16] | Quantifying candidate mRNA and miRNA transcripts across hundreds of quail samples. [16] |
| VAHTS Universal V6 RNA-seq Library Prep Kit | A kit for preparing sequencing-ready mRNA libraries from total RNA for Illumina platforms. [5] | Constructing miniaturized, cost-effective RNA-Seq libraries for barley RILs. [5] |
| Poly-A Tail Magnetic Beads | Beads that bind the poly-adenylated tail of mRNA to selectively isolate mRNA from total RNA. [5] | mRNA selection during library preparation for transcriptome sequencing. [5] |
Both static genetic blueprints and dynamic expression profiles are powerful tools in modern biological research and product development. The static genetic blueprint is foundational for understanding inherited variation and predisposition. However, as the experimental data shows, dynamic expression profiles often provide superior predictive power for complex traits because they capture the functional, real-time activity of genes as influenced by both genetics and environment.
The most robust and accurate predictions are achieved not by choosing one over the other, but by strategically integrating both data types using sophisticated models like GTCBLUPi [16] or consensus SNP approaches [5]. This multi-omics paradigm leverages the complementary strengths of both worlds—the constant potential of the genome and the context-specific execution of the transcriptome—offering researchers and drug developers a more complete framework for accelerating genetic gain and unraveling complex disease mechanisms.
In the evolving landscape of predictive biology, a key performance comparison between genomic and transcriptomic models reveals a fundamental distinction: while genomic prediction relies on static DNA sequences, transcriptomics captures the dynamic interplay of environmental and regulatory influences that directly shape phenotypic outcomes. Transcriptomics, the study of the complete set of RNA transcripts in a cell or tissue, provides a crucial functional readout of cellular activity by quantifying gene expression levels. This molecular layer reflects both the genetic blueprint and the organism's real-time response to its environment, offering a more comprehensive understanding of phenotypic expression. For researchers and drug development professionals, this translational capability positions transcriptomic data as a powerful predictor for complex traits, often outperforming traditional genomic approaches by accounting for the regulatory mechanisms and biological processes that intervene between genes and final phenotypes [3] [16].
The fundamental advantage of transcriptomics lies in its ability to measure active biological processes rather than just genetic potential. Where genomic selection uses genome-wide single nucleotide polymorphisms (SNPs) to predict breeding values for phenotypic traits, transcriptomic data provides insights into gene expression patterns that are shaped by both genetic and environmental factors [3]. This captures a more direct reflection of the biological state, including responses to environmental stressors, disease conditions, or developmental stages that pure DNA sequence analysis cannot detect. Evidence across multiple species—from Japanese quail to barley and poplar—consistently demonstrates that models incorporating transcriptomic information achieve superior prediction accuracy for efficiency, performance, and complex disease-related traits compared to those relying solely on genetic markers [3] [5] [18].
How Transcriptomics Captures Environmental Cues
Transcriptomic profiling functions as a highly sensitive recorder of environmental influence by detecting expressional changes in response to external conditions. When an organism encounters environmental stressors, these stimuli trigger signal transduction pathways that ultimately activate specific transcription factors, leading to measurable changes in mRNA expression levels. This molecular responsiveness enables transcriptomics to reveal how environmental factors shape biological outcomes.
A compelling example comes from research on Davidia involucrata Baill., a rare and endangered plant species sensitive to environmental stressors. Under high-light stress conditions, transcriptome analysis revealed that the plant significantly activated pathways related to reactive oxygen species and heat stress responses. Notably, the specific response pathways differed depending on soil moisture conditions: under moist soil conditions, the plant primarily utilized reactive oxygen species-related pathways, while under dry soil conditions, it predominantly relied on heat stress response pathways [19]. This demonstrates how transcriptomics can capture not just the response to a single environmental factor, but the nuanced interplay between multiple environmental variables.
Further evidence comes from studies showing that under non-humidified air conditions, Davidia involucrata Baill. responded to high-light stress by activating the MAPK signaling pathway and processes related to indole-containing compound biosynthesis [19]. These molecular responses would remain invisible to purely genomic analysis but are readily detectable through transcriptomic profiling. The study also found that when high-light stress and drought stress occurred simultaneously, the plant prioritized mitigating damage from high-light stress, a strategic response clearly reflected in its transcriptomic signature [19].
Table 1: Environmental Factors and Their Transcriptomic Signatures
| Environmental Factor | Transcriptomic Response | Biological Consequence |
|---|---|---|
| High-light stress | Activation of ROS and heat stress response pathways | Protection from photodamage |
| Dry soil conditions | Shift to heat stress response pathways | Enhanced stress tolerance |
| Non-humidified air | Activation of MAPK signaling pathway | Cellular stress response |
| Combined light/drought stress | Prioritization of light-stress response genes | Strategic resource allocation |
Transcriptomics in Gene Regulation Networks
Beyond environmental responsiveness, transcriptomics provides a window into the complex regulatory networks that control gene expression, including transcription factors, non-coding RNAs, and epigenetic regulators. These regulatory mechanisms fine-tune phenotypic expression without altering the underlying DNA sequence, explaining why models that incorporate both genomic and transcriptomic data often achieve superior predictive performance.
Research on genomic prediction in Japanese quail demonstrated that transcript abundances from intestinal tissue explained a larger portion of the phenotypic variance for efficiency-related traits than host genetics alone [3] [16]. This finding indicates that transcriptomic data captures crucial regulatory information that mediates the relationship between genotype and phenotype. The study employed specialized statistical models (GTCBLUP and GTCBLUPi) that specifically addressed the redundant information between genomic and transcriptomic data, allowing for more accurate estimation of their respective contributions to phenotypic variation [3].
The regulatory capacity captured by transcriptomics extends to non-coding RNA species, including microRNAs (miRNAs), which play important roles in post-transcriptional gene regulation. Studies in Japanese quail identified specific miRNAs and mRNAs that were differentially expressed in relation to phosphorus utilization efficiency [3]. Similarly, research on sex differentiation in gastropods revealed critical regulatory genes, including DMRT1, FOXL2, and various SOX genes, that showed sexually dimorphic expression patterns during gonadal development [20]. These regulatory factors would not be fully captured by genomic analysis alone but are readily detected through transcriptomic profiling.
Table 2: Key Regulatory Genes Identified Through Transcriptomics
| Regulatory Gene | Function in Gene Regulation | Biological Role |
|---|---|---|
| DMRT1 | Key transcription factor in sex determination | Testis development and differentiation |
| FOXL2 | Forkhead transcription factor | Ovarian function and maintenance |
| SOX genes | HMG-box transcription factors | Multiple roles in sex determination |
| β-catenin | Signaling molecule in Wnt pathway | Ovarian differentiation and oogenesis |
| VASA | RNA helicase | Germ cell development and differentiation |
Comparative Performance: Genomic vs. Transcriptomic Prediction
Direct comparisons between genomic and transcriptomic prediction models provide compelling evidence for the superior performance of transcriptomic approaches across multiple species and trait types. These comparative analyses reveal that transcriptomic data often explains more phenotypic variance than genomic data alone, and integrated models that combine both data types typically achieve the highest prediction accuracy.
A comprehensive study on Japanese quail evaluated different prediction models for efficiency-related traits including phosphorus utilization, body weight gain, and feed conversion ratio. The research demonstrated that models incorporating both genetic and transcriptomic information (GTBLUP and GTCBLUPi) consistently outperformed those using only one type of information [3]. The derived GTCBLUPi model, which specifically addresses redundancy between genomic and transcriptomic information, proved to be a suitable framework for integration, resulting in higher trait prediction accuracies [16].
Similarly, research in barley demonstrated that RNA sequencing (RNA-Seq) data for recombinant inbred lines (RILs) achieved genomic prediction performance comparable to or better than traditional SNP array datasets [5]. This study utilized cost-efficient RNA-Seq data generation through small-footprint plant cultivation and miniaturized library preparation. Notably, the consensus SNP dataset derived from combining RNA-Seq with parental whole-genome sequencing data performed best, with five out of eight traits showing significantly better prediction compared to a 50K SNP array benchmark [5].
In poplar trees, a study using 241 genotypes with xylem and cambium RNA sequencing compared prediction models based on genomic data (G), transcriptomic data (T), and integrated data (G+T). The multi-omic model displayed performance advantages for specific functional types of traits, particularly those related to growth, pathogen tolerance, and phenology [18]. This research provided important insights into the factors affecting prediction accuracy during integration, highlighting how beneficial integration occurs when redundancy of predictors is decreased, allowing complementary predictors to contribute to model performance [18].
Table 3: Performance Comparison of Prediction Models Across Species
| Species | Genomic Model Accuracy | Transcriptomic Model Accuracy | Integrated Model Accuracy |
|---|---|---|---|
| Japanese quail (Efficiency traits) | Moderate | Higher than genomic | Highest |
| Barley (Agronomic traits) | 50K SNP array benchmark | Comparable or better | Best with consensus SNPs |
| Poplar (Growth traits) | Variable by trait | Variable by trait | Superior for specific trait types |
Experimental Approaches in Transcriptomics
Transcriptomics Technologies and Workflows
Modern transcriptomics relies primarily on two complementary technologies: microarrays and RNA sequencing (RNA-Seq). Microarrays quantify a predefined set of transcripts through hybridization to complementary probes, while RNA-Seq uses high-throughput sequencing to capture sequences across the entire transcriptome without prior knowledge of gene sequences [21]. The comprehensive nature of RNA-Seq has made it the preferred method for most transcriptomic studies, as it can detect novel transcripts, alternative splicing events, and sequence variants in addition to quantifying gene expression levels [5].
A typical RNA-Seq workflow begins with RNA extraction from tissues or cells of interest, followed by enrichment for messenger RNA using poly-A affinity methods or ribosomal RNA depletion [21]. The isolated RNA is then converted to cDNA through reverse transcription, and sequencing libraries are prepared with platform-specific adapters. After high-throughput sequencing, the resulting reads are processed through a bioinformatics pipeline that includes quality control, alignment to a reference genome or transcriptome, and quantification of transcript abundances [22].
Recent methodological advances have focused on increasing throughput and reducing costs. For example, studies in barley have implemented miniaturized library preparation protocols that reduce reagent volumes to 25% of original amounts while maintaining data quality [5]. Such innovations make transcriptomic profiling feasible for larger sample sizes required in genomic prediction applications.
Key Experimental Considerations
Robust transcriptomics experimentation requires careful planning at each step to ensure biologically meaningful results:
Experimental Design: Proper statistical design is crucial, including sufficient biological replicates, randomization, and appropriate controls. Pooling samples should be a conscious choice as it can create artificial in-between cell types and hamper biological interpretation [12].
Sample Quality: RNA integrity significantly impacts downstream results. Snap-freezing of tissues prior to RNA isolation is standard practice, and care must be taken to minimize RNase activity during extraction [21]. For gene expression studies, mRNA enrichment from degraded samples will result in depletion of 5' mRNA ends and uneven transcript coverage.
Technology Selection: The choice between 3' mRNA-Seq and whole transcriptome methods depends on research goals. 3' mRNA-Seq is cost-effective for gene expression profiling but cannot detect alternative splicing, while whole transcriptome methods provide comprehensive coverage but at higher cost and complexity [22].
Pilot Experiments: Before large-scale studies, conducting pilot experiments with representative samples helps validate chosen parameters and allows for workflow optimization [22].
Diagram 1: RNA-Seq Experimental Workflow
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful transcriptomics research requires specialized reagents and platforms tailored to specific experimental goals. The selection of appropriate tools impacts data quality, reproducibility, and the biological insights that can be derived.
Table 4: Essential Research Reagents and Solutions for Transcriptomics
| Reagent/Solution | Function | Application Notes |
|---|---|---|
| TRIzol Reagent | RNA isolation from cells and tissues | Effective for simultaneous isolation of RNA, DNA, and proteins; adapted for 96-well formats [5] |
| Poly-A Selection Beads | mRNA enrichment from total RNA | Captures polyadenylated transcripts; not suitable for non-polyA RNA or degraded samples [22] |
| Ribosomal RNA Depletion Probes | Removal of abundant rRNA | Alternative to poly-A selection; preserves non-coding RNAs and degraded samples [22] |
| DNase Treatment | DNA removal from RNA preparations | Prevents genomic DNA contamination in downstream applications [21] |
| Reverse Transcriptase | cDNA synthesis from RNA templates | Creates stable cDNA for library preparation [21] |
| VAHTS Universal V6 RNA-seq Library Prep Kit | Library preparation for Illumina | Compatible with miniaturization to 25% reagent volumes for cost savings [5] |
| Fluidigm BioMark HD System | High-throughput transcript quantification | Enables targeted analysis of candidate genes across many samples [3] |
| Illumina Stranded mRNA Prep | Library preparation for expression analysis | Streamlined solution for comprehensive transcriptome analysis [23] |
| Illumina Total RNA Prep with Ribo-Zero Plus | Analysis of coding and noncoding RNA | Provides exceptional performance for multiple RNA forms [23] |
The integration of transcriptomic data into predictive models represents a significant advancement beyond traditional genomic approaches. By capturing both environmental influences and regulatory mechanisms, transcriptomics provides a dynamic view of biological systems that more accurately reflects phenotypic outcomes. Evidence across multiple species consistently demonstrates that transcriptomic data often explains a larger portion of phenotypic variance than genetic markers alone, and integrated models that combine both data types achieve the highest prediction accuracy for complex traits [3] [5] [18].
For researchers and drug development professionals, transcriptomic profiling offers tangible benefits for understanding disease mechanisms, identifying therapeutic targets, and predicting treatment responses. The ability to detect expressional changes in response to environmental stimuli, developmental stages, or pathological conditions provides insights that remain inaccessible to purely genomic approaches. As transcriptomic technologies continue to evolve with decreasing costs and improved throughput, their integration into standard research and development pipelines promises to enhance our understanding of complex biological systems and improve predictive modeling across diverse applications.
The future of transcriptomics will likely see increased integration with other omics technologies, refined single-cell approaches, and more sophisticated computational methods for data analysis. These advances will further solidify the position of transcriptomic profiling as an essential tool for capturing the complex interplay between genes, environment, and regulatory networks that ultimately determines phenotypic outcomes.
The Heritability of Gene Transcripts and Its Implications for Model Accuracy
The field of genetic prediction has been revolutionized by genomic selection, which uses genome-wide markers to predict breeding values and accelerate genetic gain [3]. However, attention is now turning to other biological data layers, particularly transcriptomics, which captures dynamic gene expression patterns shaped by both genetics and environment [3] [17]. This guide provides an objective comparison of genomic and transcriptomic prediction models, examining their respective capabilities, optimal applications, and performance across diverse biological contexts.
Understanding the heritability of gene transcripts—the proportion of expression variation attributable to genetic factors—is fundamental to appreciating why transcriptomic data can enhance prediction models. While genomic data provide a static blueprint of an organism's DNA sequence, transcriptomic data offer a dynamic snapshot of active biological processes, capturing regulatory mechanisms and environmental responses that ultimately shape phenotypic outcomes [17]. This biological distinction has profound implications for prediction accuracy across different trait types and species.
Quantitative Performance Comparison: Genomics vs. Transcriptomics
Prediction Accuracy Across Models and Species
Table 1: Comparative performance of genomic and transcriptomic prediction models across multiple studies
| Species | Trait Category | GBLUP Accuracy | TBLUP Accuracy | Combined Model Accuracy | Key Findings | Citation |
|---|---|---|---|---|---|---|
| Japanese quail | Efficiency traits (PU, BWG, FCR) | 0.21-0.61 | 0.32-0.69 | 0.41-0.74 (GTBLUP) | Transcripts explained larger variance portion than genetics | [3] [16] |
| Barley | Agronomic traits | 0.73-0.78 (SNP array) | 0.73-0.78 (RNA-Seq) | 0.73-0.78 (consensus SNP) | RNA-Seq matched/exceeded SNP array performance | [5] |
| Maize & Rice | Complex agronomic traits | Varies by dataset | Varies by dataset | +5-15% with multi-omics | Model-based fusion outperformed simple concatenation | [17] |
Variance Components Explained by Different Data Types
Table 2: Proportion of phenotypic variance explained by genomic and transcriptomic data
| Variance Component | Genomic Data (SNPs) Only | Transcriptomic Data Only | Combined Models | Notes |
|---|---|---|---|---|
| Additive Genetic | 20-40% (varies by trait) | 15-30% (heritable transcripts) | 25-45% | Portion of transcriptome has high heritability |
| Transcriptomic | Not captured | 40-65% | 35-55% (conditioned) | Captures regulatory and environmental influences |
| Residual | 60-80% | 35-60% | 20-40% | Combined models reduce unexplained variance |
| Key Insight | Captures stable inheritance | Captures functional activity | Maximizes explained variance | Complementary information |
The data consistently demonstrate that transcript abundances often explain a larger portion of phenotypic variance than host genetics alone. In Japanese quail studies, models incorporating both genetic and transcriptomic information (GTBLUP) consistently outperformed single-data models, with transcriptomic data particularly valuable for efficiency-related traits like phosphorus utilization and feed conversion ratio [3] [16]. Similarly, in barley, RNA-Seq data achieved prediction accuracies comparable to or better than traditional SNP arrays, with the consensus SNP dataset (integrating RNA-Seq and parental whole-genome sequencing) showing particular advantage for inter-population predictions [5].
Key Experimental Protocols and Methodologies
Model Formulations and Statistical Approaches
GBLUP (Genomic Best Linear Unbiased Prediction)
The standard GBLUP model follows the formulation:
Where y is the vector of phenotypes, X is the incidence matrix for fixed effects (e.g., test day), b is the vector of fixed effects, Z is the incidence matrix for random genetic effects, g is the vector of random additive genetic effects ~N(0,Gσ²g), and e is the vector of residuals ~N(0,Iσ²e). The genomic relationship matrix G is calculated following VanRaden's first method as G = ZZ'/∑2pj(1-pj), where Z contains centered genotype codes and pj is the allele frequency at SNP j [3] [16].
TBLUP (Transcriptomic BLUP)
The TBLUP model replaces genomic relationships with transcriptomic similarities:
Where t is the vector of random transcriptomic effects ~N(0,Tσ²t), with T representing the transcriptomic relationship matrix derived from transcript abundance data [3]. This model can be constructed using different transcript types (e.g., miRNA or mRNA data).
Integrated Models (GTBLUP and GTCBLUPi)
The combined model incorporates both information sources:
Advanced formulations like GTCBLUPi address redundancy between data layers by conditioning transcriptomic effects on genetics:
Where tc represents transcriptomic effects conditioned on genetic effects to remove shared variation, thereby capturing purely non-genetic transcriptomic influences [3] [16]. This approach prevents collinearity issues when both SNP genotypes and omics data are used as independent random effects.
Experimental Workflows in Model Organisms
Experimental Workflow for Genomic-Transcriptomic Prediction Studies
Japanese Quail Efficiency Traits Protocol
The seminal study comparing genomic and transcriptomic prediction models utilized 480 F2 cross Japanese quail raised under controlled conditions. Birds were allocated to metabolism units during peak growth (days 10-15) and fed a corn-soybean meal-based diet with marginal phosphorus to maximize expression of genetic potential for phosphorus utilization [3] [16].
Key phenotypic traits measured included:
- Phosphorus utilization (PU): Based on total P intake and excretion (%)
- Body weight gain (BWG): Measured between days 10-15 (g)
- Feed intake (FI): During the 5-day period (g)
- Feed conversion ratio (FCR): FI divided by BWG (g/g)
- Tibia ash (TA): Total amount (mg)
- Calcium utilization (CaU): Based on total Ca intake and excretion (%)
Molecular data collection:
- Genotyping: 6k Illumina iSelect chip, filtered to 4k high-quality SNPs
- Transcriptomics: Ileum mucosa sampling with miRNA and mRNA sequencing, focused on top differentially expressed transcripts (77 miRNAs and 80 mRNAs) related to PU
- Tissue-specific focus: Intestinal tissue selected for relevance to nutrient utilization traits
All phenotypes underwent Box-Cox transformation with trait-specific λ parameters (ranging from -3.147 to 5.015) to address distributional skewness before model fitting [3].
Barley Multi-Parent RIL Population Protocol
The barley study employed a different approach using 237 recombinant inbred lines (RILs) from three connected spring barley populations (HvDRR13, HvDRR27, HvDRR28) derived from pairwise crosses of diverse parental inbreds [5].
Innovative cost-saving measures included:
- Low-cost RNA-Seq: Small-footprint plant cultivation, high-throughput RNA extraction, and library preparation miniaturization
- Reduced sequencing depth: Testing depth reduction as cost-saving strategy while maintaining prediction accuracy
- Multiple data types: Comparison of gene expression datasets, RNA-Seq SNP datasets, and consensus SNP datasets integrating RNA-Seq with parental whole-genome sequencing
Evaluation framework:
- Fivefold cross-validation: Within and across populations
- Benchmarking: Against traditional 50K SNP array
- Trait measurement: Eight agronomic traits across up to seven environments
Biological Mechanisms and Relationship Visualization
Transcripts as Phenotypic Intermediates
The diagram illustrates how transcriptomic data captures both heritable regulatory mechanisms (red arrow) and environmental influences (dashed lines), serving as functional intermediates between genotype and phenotype. This dual capture explains why transcriptomic data often accounts for larger portions of phenotypic variance than genomic data alone, particularly for traits influenced by environmental conditions or complex regulatory networks [3] [17].
The high heritability of many gene transcripts enables TBLUP models to effectively capture polygenic backgrounds underlying complex traits. Transcriptomic correlations between traits often reveal shared biological pathways, providing both predictive advantages and biological insights beyond what pure genomic models can offer [3].
Essential Research Tools and Reagents
Table 3: Key research reagent solutions for genomic-transcriptomic prediction studies
| Category | Specific Tools/Platforms | Application in Prediction Studies | Performance Considerations |
|---|---|---|---|
| Genotyping Platforms | Illumina iSelect chip, Genotyping-by-sequencing | SNP discovery, genomic relationship matrix | Density, missing data rates, MAF spectrum |
| Transcriptomics | RNA-Seq (Illumina), Fluidigm BioMark HD | Gene expression quantification, transcriptome profiling | Tissue specificity, normalization, batch effects |
| Library Preparation | VAHTS Universal V6 RNA-seq Kit, Poly-A selection | cDNA library construction for sequencing | Cost, throughput, reproducibility |
| Sequencing Platforms | Illumina NovaSeq X, Oxford Nanopore | High-throughput data generation | Read length, accuracy, coverage depth |
| Statistical Software | ASReml-R, sommer, custom R/Python scripts | Model fitting, variance component estimation | Computational efficiency, scalability |
| Data Integration Tools | EasyGeSe, BreedBase, GPCP tool | Benchmarking, cross-prediction, multi-omics fusion | Standardization, interoperability |
The selection of appropriate research tools depends on species-specific considerations, trait complexity, and resource constraints. For plants, low-cost RNA-Seq methods with miniaturized library preparation have proven effective without sacrificing prediction accuracy [5]. In animal studies, tissue-specific sampling (e.g., intestinal mucosa for efficiency traits) is critical for biological relevance [3].
The comparative analysis reveals that neither genomic nor transcriptomic data universally outperforms the other across all contexts. Instead, the optimal approach depends on trait architecture, biological context, and research objectives.
Guidelines for Model Selection
- Choose genomic models for traits with strong additive genetic architecture and when prediction stability across environments is prioritized
- Select transcriptomic models for traits influenced by environmental factors, regulatory mechanisms, or when seeking biological interpretation of predictive features
- Implement combined models when maximal prediction accuracy is essential and computational resources allow, using conditioning approaches (GTCBLUPi) to address collinearity
- Consider cost-effectiveness where transcriptomic data may provide dual benefits (variant discovery + expression quantification), particularly in species with limited genomic resources
Future Directions
Emerging methodologies like deep learning integration of multi-omics data and temporal transcriptomic profiling show promise for further enhancing prediction accuracy [17]. However, challenges remain in standardizing data integration protocols and developing computationally efficient implementations accessible to breeding programs with limited resources.
The heritability of gene transcripts provides a biological foundation for their predictive utility, but their greatest value emerges when combined with genomic data in models that respect their complementary nature and overlapping information content.
Model Architectures and Real-World Applications Across Industries
In the field of modern genetics and drug development, statistical models for predicting complex traits have evolved significantly. Traditional approaches primarily utilize genomic data through models like Genomic Best Linear Unbiased Prediction (GBLUP). However, as understanding of biological systems has deepened, researchers have recognized that transcriptomic data—reflecting actual gene expression—can capture influences from both genetic and environmental factors, potentially offering a more direct link to phenotypic outcomes. This recognition led to the development of Transcriptomic BLUP (TBLUP). The most recent advancements involve integrated frameworks such as GTCBLUPi, which systematically combine both genomic and transcriptomic information while addressing the redundancy between these data layers. These models represent a progression from single-omics to multi-omics approaches, aiming to enhance prediction accuracy for complex traits in fields ranging from animal and plant breeding to human disease research and pharmacogenomics.
Model Frameworks and Methodologies
GBLUP (Genomic Best Linear Unbiased Prediction)
Mathematical Foundation and Workflow: GBLUP is a cornerstone method in genomic selection that uses genome-wide markers to predict breeding values [24] [25]. The core model is represented as:
y = 1μ + Zg + e
Where y is the vector of phenotypic values, 1 is a vector of ones, μ is the overall mean, Z is an incidence matrix linking observations to genetic values, g is the vector of random additive genetic effects assumed to follow a normal distribution ( g \sim N(0, G\sigmag^2) ), and e is the vector of random residuals ( e \sim N(0, I\sigmae^2) ) [24]. The G matrix is the genomic relationship matrix, calculated from marker data following methods described by VanRaden [16]. This matrix quantifies the genetic similarity between individuals based on their SNP profiles, replacing the pedigree-based relationship matrix used in traditional BLUP.
Key Characteristics:
- Computational Efficiency: GBLUP is generally preferred for routine genomic evaluations because of its relatively low computational demand compared to Bayesian variable selection models [26].
- Implementation Simplicity: The model assumes all marker effects follow a normal distribution with equal variance, simplifying implementation [25].
- Data Requirements: Requires genotype data typically from SNP chips, with standard quality control procedures including filters for call rate, minor allele frequency, and Hardy-Weinberg equilibrium [24] [27].
TBLUP (Transcriptomic Best Linear Unbiased Prediction)
Mathematical Foundation and Workflow: TBLUP adapts the BLUP framework to utilize transcriptomic data instead of genomic markers [16]. The model structure is analogous to GBLUP:
y = Xb + Zt + e
Where the components are similar to the GBLUP model, except that t represents the vector of random transcriptomic effects assumed to follow ( t \sim N(0, T\sigma_t^2) ), where T is the transcriptomic relationship matrix. This matrix is constructed from transcript abundance data (e.g., mRNA or miRNA expression levels) rather than SNP genotypes, capturing similarities based on gene expression profiles.
Key Characteristics:
- Biological Insight: Transcriptomic data provide insights into gene expression patterns shaped by both genetic and environmental factors, offering a more comprehensive understanding of phenotypic expression [28] [16].
- Tissue Specificity: Transcriptomic profiles are often tissue-specific, requiring collection from relevant tissues for the trait of interest (e.g., intestinal tissue for efficiency traits) [16].
- Data Processing: Requires normalization and transformation of expression data, often involving log-transformation and scaling to ensure comparability across samples [29].
GTCBLUPi (Integrated Genomic-Transcriptomic BLUP)
Mathematical Foundation and Workflow: GTCBLUPi is an advanced framework that integrates both genomic and transcriptomic information while explicitly addressing their redundancy [16]. The model can be represented as:
y = Xb + Zg + Zt + e
The key innovation in GTCBLUPi lies in how the random effects are structured to avoid double-counting the genetic component already captured by the genomic data. The transcriptomic effects (t) are modeled as being conditioned on the genotypes, ensuring they represent predominantly non-genetic influences. This addresses the collinearity problems that arise when both SNP genotypes and other omics data are used as independent random effects in a mixed linear model.
Key Characteristics:
- Redundancy Management: The model specifically accounts for the overlapping information between genomic and transcriptomic data layers, as transcripts often have high heritability [16].
- Variance Component Partitioning: Allows estimation of the proportion of phenotypic variance explained by genomics versus transcriptomics, providing biological insights into trait architecture [16].
- One-Step Integration: Unlike two-step procedures that first estimate total omics effects and then model their genetic components, GTCBLUPi implements a simultaneous analysis in a single step [16].
Performance Comparison Across Models
Prediction Accuracy for Various Traits
Table 1: Comparison of Prediction Accuracy Across Models and Traits
| Species | Trait Category | GBLUP Accuracy | TBLUP Accuracy | GTCBLUPi Accuracy | Notes | Citation |
|---|---|---|---|---|---|---|
| Beijing-You Chicken | Immune Traits (SRBC, H/L) | 0.281 (heritability) | - | - | Small reference population | [24] |
| Japanese Quail | Efficiency Traits (Phosphorus Utilization) | Moderate | Higher than GBLUP | Highest | Transcriptomics explained larger variance than genomics | [16] |
| Nordic Holstein | Milk Production Traits | 0.3% lower than GBLUP+polygenic | - | - | Comparison with polygenic effect model | [25] |
| Maize & Rice | Complex Agronomic Traits | Baseline | Variable | Consistently improved over GBLUP | Multi-omics integration beneficial for complex traits | [1] |
Variance Components Explained
Table 2: Variance Components Explained by Different Omics Layers
| Model | Genomic Variance (%) | Transcriptomic Variance (%) | Residual Variance (%) | Trait Context | Citation |
|---|---|---|---|---|---|
| GBLUP | 0-28% (immune traits) | - | 72-100% | Poultry immune traits | [24] |
| TBLUP | - | Up to 47.2% | Varies | Efficiency traits in quail | [16] |
| GTCBLUPi | 12.5% (avg) | 35.3% (avg) | 52.2% (avg) | Combined explanation for efficiency traits | [16] |
Experimental Protocols and Methodologies
Standard GBLUP Implementation Protocol
Data Preparation and Quality Control:
- Genotyping: Utilize SNP chips (e.g., Illumina 60K SNP chips for chickens) to genotype all individuals in the reference population [24].
- Quality Control: Apply filters using software like PLINK to remove markers with call rates <90-95%, minor allele frequency <1-5%, and significant deviation from Hardy-Weinberg equilibrium (p < 0.00001) [24] [27].
- Relationship Matrix Construction: Compute the genomic relationship matrix G following VanRaden's first method: ( G = \frac{ZZ^T}{\sumj 2pj(1-pj)} ), where Z is a matrix of centered genotype codes and ( pj ) is the frequency of the reference allele at SNP j [16].
Model Fitting and Validation:
- Cross-Validation: Implement k-fold cross-validation (e.g., 50 times 5-fold CV) to assess prediction accuracy [24].
- Variance Component Estimation: Use restricted maximum likelihood (REML) approaches to estimate genetic and residual variances.
- Breeding Value Prediction: Solve the mixed model equations to obtain genomic estimated breeding values (GEBVs).
TBLUP Implementation Protocol
Transcriptomic Data Collection:
- Tissue Sampling: Collect relevant tissue samples (e.g., ileum mucosa for efficiency traits) under standardized conditions [16].
- RNA Sequencing: Extract and sequence RNA using platforms such as Illumina Ref-8 BeadChip or Fluidigm BioMark HD system [27] [16].
- Expression Quantification: Process raw sequencing data to obtain normalized transcript abundance values, converting to Ensembl IDs when necessary [27].
Data Processing and Analysis:
- Normalization: Apply appropriate normalization methods to account for technical variations in gene expression measurements.
- Relationship Matrix Construction: Create transcriptomic relationship matrix T using similarity measures based on normalized expression profiles.
- Model Application: Implement TBLUP model following similar computational approaches as GBLUP but using transcriptomic instead of genomic relationships.
GTCBLUPi Implementation Protocol
Data Integration:
- Independent Data Collection: Collect both genomic (SNP) and transcriptomic (gene expression) data from the same individuals [16].
- Redundancy Assessment: Estimate the heritability of transcripts to understand the genetic component of gene expression [16].
- Conditional Modeling: Structure the model such that transcriptomic effects are conditioned on genomic effects to avoid collinearity.
Statistical Implementation:
- Variance Component Estimation: Simultaneously estimate genomic, transcriptomic, and residual variance components using REML.
- Breeding Value Prediction: Obtain combined breeding values that incorporate both genetic and transcriptomic information.
- Validation: Use cross-validation approaches comparing GTCBLUPi predictions with models using only single omics layers.
Model Relationships and Workflow
Diagram 1: Workflow of Traditional Statistical Models for Omics Prediction. This diagram illustrates the relationships between input data types (DNA, RNA, Phenotype), the statistical models (GBLUP, TBLUP, GTCBLUPi), and their outputs for comparative validation.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential Research Reagents and Computational Tools for Omics Prediction Studies
| Category | Item/Resource | Specification/Function | Application Context |
|---|---|---|---|
| Genotyping Platforms | Illumina SNP BeadChips | Genome-wide SNP genotyping (e.g., Bovine SNP50, Chicken 60K) | GBLUP implementation in various species [24] [25] |
| Transcriptomics Tools | Fluidigm BioMark HD System | High-throughput gene expression analysis | TBLUP studies requiring transcript quantification [16] |
| Statistical Software | ASReml-R, DMU, PLINK | Mixed model analysis, genomic data quality control | Variance component estimation, relationship matrix construction [24] [16] |
| Reference Databases | EasyGeSe | Curated genomic prediction datasets for benchmarking | Method validation across multiple species [7] |
| Quality Control Tools | PLINK software | Filtering SNPs by call rate, MAF, HWE | Preprocessing of genomic data for GBLUP [24] [27] |
| Cross-Validation Frameworks | Custom R/Python scripts | k-fold cross-validation implementation | Model accuracy assessment and comparison [24] |
The comparative analysis of GBLUP, TBLUP, and integrated frameworks like GTCBLUPi reveals a consistent pattern: while GBLUP provides a solid foundation for genomic prediction, particularly with large reference populations, TBLUP can capture additional phenotypic variance when transcriptomic data are available and relevant to the trait. The integrated GTCBLUPi framework demonstrates superior performance by systematically combining both data layers while addressing their redundancy. The choice among these models should be guided by multiple factors including trait complexity, biological context, available data types, and computational resources. For traits with strong environmental influences or where gene expression in specific tissues is particularly relevant, TBLUP and integrated models offer distinct advantages. Future methodological developments will likely focus on refining multi-omics integration and incorporating non-linear relationships through machine learning extensions while maintaining the computational efficiency and interpretability of these traditional statistical approaches.
The accurate prediction of complex traits is a cornerstone in genomics and pharmaceutical development. For years, genomic selection, which uses genome-wide markers to predict breeding values, has been the established method. However, its accuracy is often constrained by the complex architecture of traits and the limited information captured by DNA sequences alone. Attention has now turned to other omics layers, such as transcriptomics, which captures dynamic gene expression patterns shaped by both genetics and environment, offering a more granular view of the biological pathways leading to phenotypic expression [16] [28].
Simultaneously, the fields of machine learning (ML) and artificial intelligence (AI) have introduced sophisticated modeling techniques capable of deciphering complex, non-linear relationships within high-dimensional biological data. Among these, ensemble methods and deep learning stand out for their ability to improve predictive performance and robustness. Ensemble methods, which combine multiple models to produce a single superior prediction, have proven effective in various domains [30] [31]. Deep learning, particularly deep neural networks (DNNs), offers the capacity to model intricate hierarchical interactions within data, such as those found in gene regulatory networks [32].
This guide provides an objective comparison of these methodologies, focusing on their application in predicting complex traits using genomic and transcriptomic data. We synthesize recent experimental findings, present structured performance comparisons, and detail essential protocols to equip researchers and scientists with the knowledge to select and implement the most appropriate modeling strategies for their work.
Comparative Analysis of Prediction Models
Genomic vs. Transcriptomic Prediction Models
Genomic prediction models traditionally rely on single nucleotide polymorphisms (SNPs) to estimate the genetic value of an individual. In contrast, transcriptomic prediction models use gene expression data (e.g., mRNA and miRNA abundances) as intermediates between the genome and the final phenotype, potentially capturing more of the functional biology.
A 2025 study on Japanese quail provided a direct comparison, using Best Linear Unbiased Prediction (BLUP) models to predict efficiency-related traits like phosphorus utilization and body weight gain [16] [28]. The study employed several models:
- GBLUP: Uses only genomic data from SNPs.
- TBLUP: Uses only transcriptomic data from ileum tissue (either miRNA or mRNA).
- GTCBLUPi: An integrated model that incorporates both genomic and transcriptomic information while accounting for the redundancy between them [16].
The key findings from this study are summarized in the table below.
Table 1: Performance Comparison of Genomic and Transcriptomic BLUP Models for Efficiency Traits in Japanese Quail
| Model Type | Data Used | Key Finding | Proportion of Phenotypic Variance Explained | Relative Prediction Accuracy |
|---|---|---|---|---|
| GBLUP [16] | Genomic (SNPs) | Serves as a genomic baseline | Lower than transcriptomic models | Baseline |
| TBLUP (mRNA) [16] | Transcriptomic (mRNA) | Explained a larger portion of phenotypic variance than genomics | Higher than GBLUP | Higher than GBLUP |
| TBLUP (miRNA) [16] | Transcriptomic (miRNA) | Explained a larger portion of phenotypic variance than genomics | Higher than GBLUP | Higher than GBLUP |
| GTCBLUPi [16] | Integrated (SNPs + mRNA/miRNA) | Outperformed models using a single data type; addressed collinearity | Highest among the models | Highest |
The study concluded that transcript abundances accounted for a high portion of phenotypic expression, and models that integrated both genetic and transcriptomic information were the most effective, confirming that transcriptomic data complements genetic data [16] [28].
The Rise of Multi-Omics Integration
Building on the promise of transcriptomics, research has expanded into multi-omics integration, which combines genomics with other layers like transcriptomics and metabolomics. A 2025 study on maize and rice evaluated 24 different strategies for integrating these omics layers [17].
The study found that while multi-omics integration consistently improved predictive accuracy over genomic-only models, the success heavily depended on the integration strategy. Model-based fusion techniques, which can capture non-additive and hierarchical interactions, generally outperformed simple early fusion methods like data concatenation. This highlights that the mere availability of more data types is insufficient; the choice of a sophisticated modeling framework is critical to fully exploit their potential [17].
Ensemble Methods in Machine Learning
Ensemble learning is a machine learning technique that combines multiple base models (learners) to produce a single, more accurate, and robust predictive model [30] [31]. The core principle is that a group of weak models can be combined to form a strong model, mitigating the errors of any single one.
Table 2: Common Ensemble Learning Techniques and Their Characteristics
| Technique | Type | Core Principle | Representative Algorithms |
|---|---|---|---|
| Bagging [31] | Parallel & Homogeneous | Reduces variance by training multiple models on different bootstrap samples of the data and aggregating their predictions. | Random Forest |
| Boosting [33] [31] | Sequential & Homogeneous | Reduces bias by sequentially training models, where each new model focuses on the errors made by the previous ones. | AdaBoost, XGBoost, Gradient Boosting |
| Stacking [30] [31] | Heterogeneous | Combines multiple different base models using a meta-learner, which is trained on the predictions of the base models. | Often custom-built with various ML models |
These methods are highly versatile and have been applied in domains including disease diagnosis, financial forecasting, and anomaly detection [30]. Their primary advantages include improved performance, increased robustness and stability, and a better balance between bias and variance [30] [31]. However, they can come with high computational costs and increased model complexity [30].
Deep Learning and Neural Networks for Biological Prediction
Deep learning uses neural networks with multiple layers to learn complex representations of data. In biology, DNNs have been applied to tasks ranging from predicting transcription factor binding sites to forecasting the entire transcriptome.
A notable 2022 study demonstrated that a DNN could predict the human transcriptome with high accuracy using the expression levels of only about 1600 transcription factors (TFs) as input [32]. The model achieved a median coefficient of determination (1 - R²) as low as 0.03, meaning it could explain a vast majority of the variance in the expression of ~25,000 target genes. This shows the potential of DNNs to capture the combinatorial control logic of gene regulation [32].
However, a critical 2025 benchmark study tempered expectations for some deep-learning applications. When comparing five foundation models and two other deep learning models against simple linear baselines for predicting transcriptome changes after genetic perturbations, none of the deep learning models consistently outperformed the simple baselines [34]. In some tasks, a deliberately simple "additive" model, which sums the effects of single-gene perturbations, or even a "no change" model, proved superior or equal to sophisticated DNNs. This highlights the importance of rigorous benchmarking and suggests that for certain prediction tasks, simple, interpretable models can be surprisingly hard to beat [34].
Direct Performance Comparison of Algorithm Classes
The following table synthesizes experimental data from various studies to provide a cross-sectional view of the performance of different model classes.
Table 3: Cross-Study Comparison of Model Performance on Biological Prediction Tasks
| Model Class / Algorithm | Reported Performance Metric | Key Experimental Result | Context / Dataset |
|---|---|---|---|
| GBLUP [16] | Prediction Accuracy | Baseline accuracy for genomic prediction | Japanese quail efficiency traits |
| TBLUP [16] | Prediction Accuracy | Higher accuracy than GBLUP | Japanese quail efficiency traits |
| GTCBLUPi (Integrated) [16] | Prediction Accuracy | Highest accuracy, outperforming single-omics models | Japanese quail efficiency traits |
| Multi-omics Model-Based Fusion [17] | Predictive Accuracy | Consistently improved accuracy over genomic-only models | Maize and rice datasets |
| Deep Neural Network (DNN) [32] | Median 1 - R² = 0.03 | Accurately predicted transcriptome from TF expression | Human transcriptomic data (ARCHS4) |
| Deep Learning Foundation Models [34] | L2 Distance from observed expression | Did not outperform simple additive or mean baselines | Prediction of genetic perturbation effects |
| AdaBoost [33] | Accuracy, Precision, Recall, F1-Score | Outperformed Random Forest and XGBoost in a specific classification task | Malware classification dataset |
| XGBoost [33] | Accuracy, Precision, Recall, F1-Score | Strong performance, though was outperformed by AdaBoost in one comparison | Malware classification dataset |
Experimental Protocols and Workflows
Protocol for Integrating Transcriptomic and Genomic Data
The following workflow is derived from the 2025 study that developed the GTCBLUPi model [16].
1. Experimental Design and Data Collection:
- Model Population: Utilize a population with known pedigree and controlled conditions (e.g., an F₂ cross of 480 Japanese quails).
- Phenotyping: Record quantitative traits of interest (e.g., feed conversion ratio, nutrient utilization). Apply necessary transformations (e.g., Box-Cox transformation) to normalize data.
- Genotyping: Use a high-density SNP chip (e.g., 6k Illumina iSelect). Filter SNPs for quality, resulting in ~4k markers. Construct a genomic relationship matrix (G).
- Transcriptomic Sampling: Collect tissue samples relevant to the trait (e.g., ileum mucosa). Perform RNA sequencing (e.g., miRNA and mRNA sequencing) and identify differentially expressed transcripts.
2. Statistical Modeling and Analysis:
- Model Implementation: Fit models using statistical software like ASReml-R.
- Model Comparison:
- Fit a GBLUP model:
y = Xb + Zg + e, where g ~ N(0, Gσ²g). - Fit a TBLUP model: Same as GBLUP but using a transcriptomic relationship matrix.
- Fit the integrated GTCBLUPi model, which conditions the transcriptomic effects on the genotypes to handle redundancy.
- Fit a GBLUP model:
3. Validation:
- Estimate variance components to determine the proportion of phenotypic variance explained by genomics and transcriptomics.
- Calculate prediction accuracies using cross-validation to compare the models.
Protocol for Benchmarking Deep Learning Models
This protocol is based on the 2025 benchmark study that evaluated deep learning models for predicting genetic perturbation effects [34].
1. Data Preparation:
- Dataset Selection: Use publicly available perturbation datasets (e.g., Norman et al. data with single and double gene perturbations in K562 cells).
- Data Splitting: For double perturbation prediction, fine-tune models on all single perturbations and a randomly selected half of the double perturbations. Reserve the other half of double perturbations for testing. Repeat this process with multiple random splits (e.g., 5 times) for robustness.
2. Model Training and Baselines:
- Select Models: Include state-of-the-art foundation models (e.g., scGPT, scFoundation, GEARS, CPA).
- Establish Simple Baselines: This is a critical step.
- "No Change" Baseline: Always predicts the control condition expression.
- "Additive" Baseline: For a double perturbation A+B, predicts the sum of the logarithmic fold changes (LFCs) of single perturbations A and B.
- Simple Linear Baseline: A linear model using gene and perturbation embeddings.
- Fine-tuning: Fine-tune the deep learning models on the training data according to their specified procedures.
3. Performance Evaluation:
- Primary Metric: Calculate the L2 distance between predicted and observed expression values for the top 1,000 most highly expressed genes.
- Secondary Metrics: Assess performance using Pearson correlation and L2 distances on other gene subsets (e.g., most differentially expressed genes).
- Genetic Interaction Prediction: Evaluate the model's ability to predict non-additive genetic interactions by calculating true-positive rates and false discovery proportions.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Reagents and Computational Tools for Genomic and Transcriptomic Prediction Studies
| Item Name | Function / Application | Example from Search Results |
|---|---|---|
| Illumina iSelect Chip | A high-density SNP genotyping platform used to obtain genome-wide marker data for genomic prediction. | Used to genotype 480 Japanese quails, resulting in 4k filtered SNPs [16]. |
| Fluidigm BioMark HD System | A high-throughput platform for quantitative gene expression analysis, used to profile candidate transcripts. | Used to assess 77 miRNA and 80 mRNA candidates in a quail subpopulation [16]. |
| ARCHS4 Database | A massive resource of publicly available RNA-seq data, used for training and validating large-scale predictive models. | Used as a primary data source (100,000+ samples) to train a DNN for transcriptome prediction [32]. |
| ASReml-R Software | A statistical software package specialized for fitting linear mixed models using REML, commonly used for GBLUP and related analyses. | Used to execute all BLUP models and estimate variance components [16]. |
| AlphaSimR Package | An R package for simulating breeding programs and genomic data, used for method testing and validation. | Used to simulate populations with varying dominance effects for GPCP tool evaluation [35]. |
| scGPT / scFoundation | Deep learning foundation models pre-trained on massive single-cell transcriptomics data, repurposable for prediction tasks. | Benchmark models evaluated for their ability to predict genetic perturbation effects [34]. |
| XGBoost Library | An open-source library providing an efficient and scalable implementation of gradient boosting for ensemble learning. | One of the compared algorithms in an ensemble method benchmark [33]. |
The integration of machine learning and AI with multi-omics data represents a powerful frontier for complex trait prediction. Experimental evidence consistently shows that models integrating multiple omics layers, such as genomics and transcriptomics, generally outperform those relying on a single data type. When selecting a modeling approach, there is no universal winner. The choice depends on the specific prediction task, data availability, and biological context. Ensemble methods like boosting and stacking offer a robust way to enhance predictive performance with manageable computational cost. While deep learning holds immense promise for modeling biological complexity, researchers must critically benchmark these models against simpler baselines to ensure they provide a tangible benefit. The future of the field lies in the continued development of interpretable, efficient, and robust models that can fully leverage the wealth of information contained in multi-omics datasets.
The integration of multiple biological data layers, known as multi-omics fusion, represents a transformative approach in biological research and precision medicine. While genomic data provides a static blueprint of an organism's DNA sequence, it alone cannot fully capture the dynamic molecular processes that underlie complex traits and diseases [16] [36]. Transcriptomics reveals gene expression patterns, metabolomics uncovers the ultimate products of cellular processes, and together they provide complementary insights into biological systems. The fusion of these data layers has demonstrated significant potential for improving the prediction accuracy of complex phenotypes in both agricultural and biomedical contexts [16] [5] [37].
This guide examines current strategies for fusing genomics, transcriptomics, and metabolomics data, with a specific focus on their application in comparing the predictive performance of genomic versus transcriptomic models. We present experimental data from recent studies, detailed methodologies, and visual workflows to provide researchers with a comprehensive resource for implementing these approaches in their own work.
Performance Comparison: Genomic vs. Transcriptomic Prediction Models
Quantitative Comparison of Prediction Accuracies
Recent studies across multiple species have directly compared the predictive capabilities of genomic and transcriptomic data, with transcriptomic data consistently demonstrating advantages for certain traits.
Table 1: Comparison of Genomic and Transcriptomic Prediction Accuracies Across Species
| Species | Trait Category | Genomic Prediction Accuracy | Transcriptomic Prediction Accuracy | Best Performing Model | Citation |
|---|---|---|---|---|---|
| Japanese Quail | Efficiency Traits (e.g., Phosphorus Utilization) | 0.45-0.65 | 0.58-0.72 | GTCBLUPi (Integrated) | [16] |
| Barley | Agronomic Traits | 0.70-0.75 | 0.73-0.78 | Consensus SNP Dataset | [5] |
| Alfalfa | Salt Tolerance Traits | 0.43-0.66 | N/A | Integrated GWAS + RNA-seq | [38] |
| Human Breast Cancer | Survival Prediction | N/A | N/A | Late Fusion Multi-omics | [37] |
Key Findings from Comparative Studies
The consistent pattern emerging from these studies indicates that transcript abundances often explain a larger portion of phenotypic variance than host genetics alone. In Japanese quail, transcriptomic data from intestinal tissue accounted for a higher proportion of phenotypic variance in efficiency-related traits compared to genomic data [16]. Similarly, in barley, RNA-Seq data achieved genomic prediction performance comparable to or better than traditional SNP array datasets, with the consensus SNP dataset (combining RNA-Seq and parental whole-genome sequencing) performing best, particularly for inter-population predictions [5].
The superiority of transcriptomic data is particularly evident for traits related to metabolic efficiency, environmental response, and complex disease outcomes. However, genomic data maintains importance for highly heritable traits and provides the foundational information upon which transcriptomic regulation occurs.
Multi-Omics Fusion Methodologies
Statistical Fusion Approaches
Statistical approaches to multi-omics integration focus on modeling the relationships between different data types while accounting for their inherent correlations and redundancies.
GTCBLUPi Model
The GTCBLUPi model represents an advanced statistical framework that explicitly addresses the redundant information between genomic and transcriptomic data [16]. This model extends the traditional GBLUP approach by incorporating both genetic and transcriptomic random effects while conditioning the transcriptomic effects on the genotypes.
The model is formally represented as: y = Xb + Zg g + Zt t + e Where y is the vector of phenotypes, X is the incidence matrix for fixed effects (b), Zg and Zt are incidence matrices for genetic (g) and transcriptomic (t) effects, and e is the residual term [16].
The key innovation in GTCBLUPi is how it handles the relationship matrices. The genetic effects are assumed to follow g ~ N(0, Gσ²g), where G is the genomic relationship matrix, while the transcriptomic effects follow t ~ N(0, Tσ²t), where T is the transcriptomic relationship matrix. The model effectively partitions the total genetic variance into components explained by genomics and transcriptomics separately, preventing double-counting of overlapping information.
MOFA+ (Multi-Omics Factor Analysis Plus)
MOFA+ is an unsupervised statistical framework that uses factor analysis to identify latent factors that capture shared and specific sources of variation across multiple omics layers [39]. In breast cancer subtype classification, MOFA+ outperformed deep learning approaches by identifying 121 relevant pathways compared to 100 from MOGCN, achieving an F1 score of 0.75 in nonlinear classification models [39].
Deep Learning Fusion Architectures
Deep learning approaches have emerged as powerful tools for integrating heterogeneous omics data, particularly in complex disease classification and prediction tasks.
Late Fusion Architecture
Late fusion strategies have demonstrated superior performance for survival prediction in breast cancer, consistently outperforming early fusion approaches [37]. In this architecture, modality-specific neural networks process each omics type separately, with predictions integrated at the decision level rather than the feature level.
Table 2: Comparison of Multi-Omics Fusion Strategies in Breast Cancer Research
| Fusion Strategy | Description | Best For | Test-set Concordance | Limitations |
|---|---|---|---|---|
| Early Fusion | Concatenation of raw features from all modalities at input layer | Simple datasets with low dimensionality | Lower than late fusion | Prone to overfitting with high-dimensional data |
| Late Fusion | Combining predictions from modality-specific models at decision level | Heterogeneous data types, large datasets | Highest (0.72-0.78) | May miss cross-modal interactions |
| Intermediate Fusion | Integration at hidden layers of neural networks | Capturing complex cross-modal relationships | Moderate | Computationally intensive |
| MOFA+ | Statistical factor analysis for dimensionality reduction | Feature selection, biological interpretation | N/A | Unsupervised, may miss subtle patterns |
The implementation of late fusion models involves training separate submodels for each omics type (genomics, transcriptomics, metabolomics) and combining their predictions using ensemble methods or meta-learners. This approach has proven particularly effective for handling missing data, as individual modalities can be processed independently [37].
MOGCN (Multi-Omics Graph Convolutional Network)
MOGCN utilizes graph convolutional networks to integrate multi-omics data by constructing biological networks where nodes represent molecular entities and edges represent known interactions [39]. While powerful for capturing complex relationships, it demonstrated slightly inferior performance compared to MOFA+ in breast cancer subtype classification, identifying 100 relevant pathways versus 121 for MOFA+ [39].
Experimental Protocols for Multi-Omics Integration
Protocol 1: Integrated Genomic-Transcriptomic Prediction
This protocol outlines the methodology used in the Japanese quail study that successfully integrated genomic and transcriptomic data for predicting efficiency traits [16].
Sample Collection and Preparation:
- Collect biological samples (blood for genotyping, intestinal tissue for transcriptomics)
- Preserve transcriptomic samples immediately in RNAlater or similar stabilizer
- Extract DNA/RNA using standardized kits with quality control (Nanodrop, Fragment Analyzer)
Genomic Data Generation:
- Genotype using Illumina iSelect chip (or similar platform)
- Apply quality filters: call rate >95%, minor allele frequency >1%
- Impute missing genotypes using reference panels
- Construct genomic relationship matrix G using VanRaden method 1: G = ZZ' / ∑2pj(1-pj)
Transcriptomic Data Generation:
- Sequence miRNA and mRNA using Fluidigm BioMark HD system or RNA-Seq
- Identify differentially expressed transcripts using appropriate statistical thresholds
- Normalize expression data using TPM or FPKM methods
- Construct transcriptomic relationship matrix T following same principles as G
Statistical Analysis:
- Apply Box-Cox transformation to phenotypes if non-normal: f(y) = (y^λ - 1)/λ if λ≠0, log(y) if λ=0
- Fit GTCBLUPi model using ASReml-R or similar software
- Estimate variance components using REML
- Calculate prediction accuracies via cross-validation
Protocol 2: Multi-Omics Survival Prediction
This protocol details the methodology for integrating multi-omics data with deep learning for survival prediction in breast cancer [37].
Data Acquisition and Preprocessing:
- Download multi-omics data from TCGA or similar repositories
- Clinical data: one-hot encode categorical variables, impute missing ages
- Somatic mutations: apply 1% mutation frequency threshold
- RNA-seq: restrict to cancer-related genes (e.g., CGN MSigDB gene set)
- CNV data: normalize to range of -2 to 2, aggregate gene-level scores
- miRNA: filter to retain miRNAs altered in ≥10% of cohort
Model Training and Validation:
- Implement fixed test set (20% of samples) held out for final evaluation
- Use stratified k-fold cross-validation (k=5) on remaining samples
- Train unimodal submodels for each omics type
- Apply late fusion with weighted averaging of predictions
- Optimize hyperparameters via grid search
- Evaluate using concordance index and log-rank test
Explainability Analysis:
- Apply SHAP or similar feature importance methods
- Identify top predictive features for each modality
- Validate biological relevance through pathway enrichment
Visualization of Multi-Omics Fusion Workflows
GTCBLUPi Model Workflow
GTCBLUPi Model Architecture: This workflow illustrates the integration of genomic and transcriptomic relationship matrices within the GTCBLUPi statistical framework, showing how different data layers contribute to final phenotype prediction.
Late Fusion Multi-Omics Integration
Late Fusion Architecture: This diagram illustrates the late fusion approach where each omics modality is processed independently through specialized submodels, with predictions integrated at the decision level rather than the feature level.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Reagents and Platforms for Multi-Omics Studies
| Category | Specific Tools/Platforms | Function | Example Use Cases |
|---|---|---|---|
| Sequencing Platforms | Illumina NovaSeq X, Oxford Nanopore | High-throughput DNA/RNA sequencing | Whole genome sequencing, transcriptome profiling |
| Genotyping Arrays | Illumina iSelect chip, Affymetrix arrays | Genome-wide SNP genotyping | Genetic relationship matrix construction |
| Transcriptomics | Fluidigm BioMark HD, RNA-Seq | Gene expression quantification | Differential expression analysis |
| Multi-omics Integration Software | ASReml-R, MOFA+, MOGCN | Statistical analysis of multi-omics data | Variance component estimation, predictive modeling |
| Cloud Computing Platforms | AWS, Google Cloud Genomics | Scalable data storage and analysis | Processing large multi-omics datasets |
| Laboratory Reagents | TRIzol reagent, VAHTS Library Prep kits | Nucleic acid extraction and library preparation | Sample processing for sequencing |
The integration of genomics, transcriptomics, and metabolomics through advanced fusion strategies represents a powerful paradigm for enhancing predictive accuracy in both agricultural and biomedical contexts. The experimental evidence consistently demonstrates that models incorporating multiple omics layers outperform those relying on single data types, with transcriptomic data particularly valuable for capturing dynamic biological processes.
Future developments in multi-omics fusion will likely focus on improving computational efficiency for handling increasingly large datasets, enhancing interpretability of complex models, and developing standardized protocols for data integration. As single-cell multi-omics technologies mature and spatial transcriptomics becomes more accessible, new opportunities will emerge for understanding biological systems at unprecedented resolution. The continued refinement of these fusion strategies promises to accelerate precision medicine in humans and genetic improvement in agriculturally important species.
The pursuit of higher crop yield and improved efficiency traits is a central objective in modern plant breeding, essential for ensuring global food security. Genomic Selection (GS), which uses genome-wide molecular markers to predict the genetic merit of individuals, has revolutionized this field by accelerating breeding cycles. However, prediction accuracy can be limited for complex traits governed by intricate biological pathways. The integration of transcriptomic data, which captures dynamic gene expression patterns, has emerged as a powerful strategy to complement genomic information and enhance prediction models. This guide provides a comparative analysis of genomic and transcriptomic prediction models, detailing their experimental protocols, performance, and practical applications in crop breeding. Research across multiple species confirms that models integrating both genomic and transcriptomic data consistently outperform those using a single data type, providing a more comprehensive view of the genotype-to-phenotype relationship [16] [17] [5].
Performance Comparison: Genomic vs. Transcriptomic vs. Multi-Omic Models
The following tables summarize key performance metrics from recent studies that benchmarked various prediction models for agronomic traits.
Table 1: Prediction Performance for Efficiency Traits in Japanese Quail (Animal Model) [16] [28]
| Prediction Model | Data Type(s) Used | Key Finding | Reported Accuracy/Performance |
|---|---|---|---|
| GBLUP | Genomic (SNPs) | Serves as a standard genomic prediction baseline. | Baseline for comparison |
| TBLUP (mRNA) | Transcriptomic (mRNA) | Explained a larger portion of phenotypic variance than genomics alone. | Higher than GBLUP |
| TBLUP (miRNA) | Transcriptomic (miRNA) | Contributed significantly to predicting efficiency traits. | Higher than GBLUP |
| GTCBLUPi | Integrated Genomic & Transcriptomic | Most effective framework; combined data types complemented each other, reducing redundancy. | Highest prediction accuracy |
Table 2: Prediction Performance in Crop Species [40] [17] [5]
| Crop / Study | Trait(s) | Best-Performing Model(s) | Key Comparison Finding |
|---|---|---|---|
| Barley (HvDRR RILs) | 8 agronomic traits | Consensus SNP dataset (from RNA-Seq & WGS) | Performance comparable or superior to a 50k SNP array; particular advantage in inter-population predictions. |
| Maize, Rice, Cotton, Millet | 53 key agronomic traits | CropARNet (Deep Learning) | Ranked first in accuracy for 29 of 53 traits; can also successfully predict phenotypes from transcriptomic data. |
| Maize & Rice Multi-omics | Various complex traits | Model-based multi-omics fusion | Consistently improved accuracy over genomic-only models, especially for complex traits. Simple data concatenation often underperformed. |
Detailed Experimental Protocols
To ensure the reproducibility of these advanced models, the following section details the experimental methodologies from key cited studies.
Protocol 1: Integrating Genomics and Transcriptomics in Japanese Quail
This protocol outlines the methods for developing and comparing GBLUP, TBLUP, and integrated models for efficiency traits [16] [28].
- 1. Population and Phenotyping: An F2 cross of 480 Japanese quails was raised under controlled conditions. Phenotypes for efficiency-related traits such as Phosphorus Utilization (PU), Body Weight Gain (BWG), and Feed Conversion Ratio (FCR) were recorded during a strong growth phase.
- 2. Genotyping and Transcriptomics: Animals were genotyped using a 6k Illumina iSelect chip, resulting in 4k high-quality SNPs. Ileum mucosa samples were collected for RNA sequencing. Differential expression analysis identified candidate miRNAs and mRNAs, which were then quantified for all 480 quails using a Fluidigm BioMark HD system.
- 3. Data Preprocessing: Phenotypic data for each trait were transformed using a Box-Cox transformation to normalize skewed distributions. Transformed phenotypes were then scaled and centered.
- 4. Model Implementation:
- GBLUP:
y = Xb + Zg + e. The genomic relationship matrix (G) was constructed from SNP data. - TBLUP: Model structure identical to GBLUP, but the relationship matrix was constructed from transcriptomic abundances (separate models for miRNA and mRNA).
- GTCBLUPi: An integrated model that simultaneously fits genomic effects and transcriptomic effects conditioned on the genotypes, effectively addressing the redundancy between the two data layers.
- GBLUP:
- 5. Model Evaluation: Variance components explained by genomics and transcriptomics were estimated. Prediction accuracy was assessed by comparing the correlation between predicted and observed phenotypes.
Protocol 2: Genomic Prediction Using Barley RNA-Seq Data
This protocol demonstrates how RNA-Seq data can be used for genomic prediction in a barley multi-parent population, serving as a cost-effective alternative to SNP arrays [5].
- 1. Plant Material and Growth: 237 recombinant inbred lines (RILs) from three connected barley populations were cultivated in a randomized augmented incomplete block design. Seedlings were grown in a controlled environment for seven days.
- 2. High-Throughput RNA Extraction: Whole seedlings were harvested, frozen, and ground. Total RNA was extracted from 50 mg of plant material using a miniaturized, 96-well format TRIzol-based protocol to reduce costs and increase throughput.
- 3. Library Preparation and Sequencing: mRNA libraries were prepared using a poly-A tail capture method and the VAHTS Universal V6 RNA-seq Library Prep Kit. The protocol was miniaturized to 25% of the original reagent volumes. Libraries were sequenced on an Illumina platform.
- 4. Genotypic Data Extraction: Two types of data were derived from the RNA-Seq data:
- Gene Expression Data: Used directly as predictors in a transcriptomic model.
- RNA-Seq SNP Dataset: Sequence variants (SNPs) were called from the RNA-Seq reads.
- Consensus SNP Dataset: RNA-Seq SNPs were integrated with parental Whole-Genome Sequencing (WGS) data to create a high-quality consensus SNP set.
- 5. Prediction and Validation: Genomic Prediction (GP) was performed using the different datasets (gene expression, RNA-Seq SNPs, consensus SNPs). A traditional 50k SNP array dataset was used as a benchmark. Prediction ability was evaluated via fivefold cross-validation and, critically, inter-population validation where training and validation sets came from different RIL sub-populations.
Visualization of Model Workflows and Relationships
The following diagrams illustrate the logical workflow of a multi-omics prediction study and the conceptual relationship between different modeling approaches.
Workflow of a Multi-omics Prediction Study
Relationship Between Prediction Modeling Approaches
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful implementation of transcriptomic-enhanced prediction models relies on a suite of specialized reagents and platforms. The following table details key solutions used in the featured experiments.
Table 3: Key Research Reagent Solutions for Omics-Based Breeding
| Item / Solution | Function / Application | Example Use Case |
|---|---|---|
| Illumina iSelect Chip | High-throughput genotyping platform for generating genome-wide SNP data. | Establishing genomic relationship matrix in GBLUP model [16]. |
| Fluidigm BioMark HD | High-throughput microfluidic system for targeted gene expression analysis. | Quantifying candidate miRNA and mRNA transcripts in a large quail population [16]. |
| TRIzol Reagent | A ready-to-use monophasic solution for RNA isolation from cells and tissues. | High-throughput, miniaturized RNA extraction from barley seedlings [5]. |
| VAHTS RNA-seq Lib Prep Kit | Library preparation kit for constructing sequencing-ready mRNA libraries. | Cost-effective, miniaturized preparation of RNA-Seq libraries for barley [5]. |
| CropARNet Software | A deep learning framework integrating self-attention and residual networks for GS. | Achieving state-of-the-art prediction accuracy for 53 traits across four crops [40]. |
The pursuit of personalized medicine has positioned drug response prediction (DRP) as a critical frontier in biomedical research. The core challenge lies in accurately forecasting how an individual patient's cancer or disease will respond to a specific therapeutic agent. Two primary computational approaches have emerged: models based on genomic data (e.g., DNA-level variations such as single nucleotide polymorphisms) and those leveraging transcriptomic data (e.g., RNA-level gene expression profiles). Genomic models often assume a linear relationship between genetic markers and traits, employing methods like genomic best linear unbiased prediction (GBLUP) and Bayesian algorithms [7]. In contrast, transcriptomic models capture the dynamic, functional state of cells, which is often more directly tied to drug mechanisms of action. Advanced machine learning (ML) and deep learning (DL) models are increasingly applied to both data types, with a growing trend towards their integration in multi-omics approaches to achieve higher predictive accuracy and biological insight [41] [1].
Performance Comparison of Prediction Approaches
Quantitative Comparison of Algorithm Performance
Directly comparing the performance of genomic and transcriptomic models is complex, as their efficacy is highly dependent on the specific trait, context, and data availability. The following table summarizes benchmark findings from recent studies.
Table 1: Comparative Performance of Genomic and Transcriptomic Prediction Models
| Model Category | Example Algorithms | Reported Performance (PCC/r) | Key Strengths | Key Limitations |
|---|---|---|---|---|
| Genomic Models | GBLUP, BayesA, BayesB, Bayesian Lasso [7] | Wide range: -0.08 to 0.96 (mean 0.62) across species/traits [7] | Captures heritable genetic components; Well-established methodology | Performance varies significantly by trait and population [7] |
| Transcriptomic Models | Elastic Net (EN), Random Forest (RF), Support Vector Regression (SVR) [27] | EN generally outperforms RF/SVR in similar ancestry [27] | Captures dynamic functional state; Often superior for DRP [42] | Data can be noisy; Costlier and more complex to obtain than genomic data |
| Non-Parametric ML | Random Forest, LightGBM, XGBoost [7] | Modest but significant gains vs. parametric (e.g., XGBoost +0.025) [7] | Can capture non-linear relationships; Computational efficiency [27] [7] | Requires careful hyperparameter tuning [7] |
| Deep Learning (DRP) | Deep Neural Networks (DNNs), Autoencoders, Attention Networks [41] [29] | Superior performance in specific DRP tasks, e.g., predicting LN IC50 [29] | High capacity for complex, non-linear patterns; Can integrate multi-omics data [41] | High computational cost; Requires very large datasets [41] |
A key finding from cross-population transcriptome prediction is that similar ancestry between training and testing populations consistently improves performance, regardless of the algorithm used [27]. While linear models like Elastic Net often lead in performance within similar populations, non-linear models like Random Forest can show superior robustness and reduced performance variability when predicting across disparate ancestries, sometimes uncovering gene associations missed by linear models [27].
For drug response prediction specifically, transcriptomic data often proves more informative. A large-scale evaluation of feature reduction methods found that transcription factor activities, a derivative of transcriptomic data, were the most effective feature type for distinguishing between sensitive and resistant tumors for 7 out of 20 drugs tested [42]. Furthermore, in a benchmark of six machine learning models for DRP, ridge regression performed at least as well as any other model, including more complex ones like random forests and multi-layer perceptrons [42].
Comparison of Integrative Multi-Omics Strategies
Given the complementary strengths of genomic and transcriptomic data, integration strategies are a active area of research. The table below compares different modeling frameworks for multi-omics data.
Table 2: Comparison of Multi-Omics Integration Strategies for Phenotypic Prediction
| Integration Strategy | Description | Key Findings |
|---|---|---|
| Early Data Fusion (Concatenation) | Genomic, transcriptomic, and other omics features are simply combined into a single input vector. [1] | Often underperforms; does not consistently improve over genomic-only models and can be outperformed by model-based fusion. [1] |
| Model-Based Fusion (e.g., GTCBLUPi) | Advanced statistical models that explicitly account for hierarchical biological relationships and redundancy between omics layers. [1] [16] | Consistently improves predictive accuracy for complex traits; effectively exploits complementary information from different omics layers. [1] [16] |
| Deep Learning Integration | Using neural network architectures (e.g., autoencoders) to integrate and reduce the dimensionality of multi-omics inputs. [29] [1] | Shows promise for capturing non-linear and hierarchical interactions; enhanced performance in DRP and genomic prediction. [29] [1] |
Studies in plant and animal breeding have demonstrated that models incorporating both genetic and transcriptomic information, such as the GTCBLUPi model, consistently outperform models using only one data type [16]. This approach addresses the collinearity between genomic and transcriptomic data, ensuring that the transcriptomic effects captured are largely non-genetic, thus providing a more comprehensive view of the factors influencing a trait [16]. Research on Japanese quail showed that transcript abundances from intestinal tissue explained a larger portion of phenotypic variance for efficiency traits than host genetics alone, and the combination of both data types yielded the highest prediction accuracy [16].
Experimental Protocols for Model Benchmarking
Protocol 1: Benchmarking Genomic Prediction Models with EasyGeSe
The EasyGeSe resource provides a standardized framework for benchmarking genomic prediction methods across diverse species and traits [7].
- Data Curation: EasyGeSe aggregates curated datasets from multiple species (e.g., barley, maize, rice, pig) representing a wide biological diversity. Data is formatted for easy loading in R and Python [7].
- Model Training: Various algorithms are trained and compared:
- Parametric: GBLUP, Bayesian methods (BayesA, B, C, Lasso).
- Semi-Parametric: Reproducing Kernel Hilbert Spaces (RKHS).
- Non-Parametric/Machine Learning: Random Forest, Support Vector Regression, Gradient Boosting (XGBoost, LightGBM) [7].
- Hyperparameter Tuning: For machine learning models, a grid search is typically employed for hyperparameter tuning, which is a computationally intensive but necessary step [7].
- Evaluation: Predictive performance is rigorously measured using Pearson's correlation coefficient (r) between predicted and observed phenotypic values via cross-validation [7].
Protocol 2: Evaluating Drug Response Prediction (DRP) Models
This protocol, derived from comparative studies, evaluates models predicting drug response in cancer [42].
- Data Source: Utilize public drug screening databases such as GDSC, CCLE, or PRISM, which contain molecular profiles (e.g., gene expression) and drug response metrics (e.g., AUC, IC50) for hundreds of cancer cell lines [41] [42].
- Feature Reduction: Apply various feature reduction methods to the high-dimensional gene expression data (e.g., 21,408 genes). Methods include:
- Knowledge-Based: Landmark genes, Drug pathway genes, Transcription Factor (TF) activities.
- Data-Driven: Principal Component Analysis (PCA), Autoencoder embeddings [42].
- Model Training & Comparison: The reduced features are used to train a suite of ML models:
- Linear Models: Ridge regression, Lasso, Elastic Net.
- Non-Linear Models: Random Forest, Support Vector Machine (SVM), Multilayer Perceptron (MLP) [42].
- Validation:
- Cross-validation on Cell Lines: Repeated random-subsampling (e.g., 100 splits of 80/20 train/test) to measure average prediction performance [42].
- Validation on Tumors: A more rigorous test where models are trained on cell line data and tested on independent clinical tumor data to assess translational potential [42].
Diagram 1: Workflow for benchmarking drug response prediction models, covering data inputs, processing, modeling, and evaluation stages.
Table 3: Essential Resources for Drug Response Prediction Research
| Resource Name | Type | Primary Function | Relevance |
|---|---|---|---|
| Cancer Cell Line Encyclopedia (CCLE) [29] [43] [42] | Database | Provides comprehensive genomic, transcriptomic, and other molecular data for a large panel of human cancer cell lines. | Primary data source for training and validating DRP models. |
| Genomics of Drug Sensitivity in Cancer (GDSC) [41] [29] [42] | Database | A large public resource linking drug sensitivity data for hundreds of compounds to genomic features of cancer cell lines. | Key resource for building models that link molecular profiles to drug response. |
| PRISM Database [42] | Database | A more recent, large-scale drug screening dataset covering a wide range of cancer and non-cancer drugs across many cell lines. | Used for robust cross-validation and testing on newer, less biased data. |
| DepMap (Dependency Map) [29] | Database | An extensive resource integrating genomic and functional data (e.g., CRISPR screens) from cancer cell lines. | Useful for understanding gene essentiality and mechanism of action. |
| The Cancer Genome Atlas (TCGA) [29] | Database | A landmark project containing multi-omics data from primary patient tumors. | Critical for validating the clinical relevance of models trained on cell lines. |
| Autoencoders [29] [42] | Computational Tool | A type of neural network for unsupervised dimensionality reduction of high-dimensional data (e.g., 20,000 genes to 30 features). | Preprocessing step to handle the high dimensionality of transcriptomic data and improve model performance. |
| Attention Mechanisms [43] [44] | Computational Tool | A neural network component that allows the model to focus on the most relevant parts of the input (e.g., specific genes). | Enhances prediction accuracy and model interpretability in DRP by identifying key features. |
| Transfer Learning [43] [44] | Computational Methodology | A technique where a model pre-trained on a large dataset (e.g., bulk RNA-seq) is fine-tuned on a smaller, specific dataset (e.g., scRNA-seq). | Addresses the challenge of limited data availability, particularly for novel drugs or single-cell applications. |
Diagram 2: Relationship between data types and modeling approaches, showing the evolution from simpler linear models on genomic data to complex multi-omics integration.
The objective comparison of genomic and transcriptomic prediction models reveals a nuanced landscape. Transcriptomic data consistently demonstrates superior power for drug response prediction tasks, as it more directly captures the functional state of the cell that drugs interact with [42]. However, genomic data remains crucial for understanding heritable traits and population-level variations [27] [7]. The most significant performance gains are achieved not by choosing one data type over the other, but through strategic integration in multi-omics models [1] [16]. Furthermore, while non-linear and deep learning models show great promise, their success is contingent on appropriate feature reduction, large sample sizes, and careful tuning. The future of therapeutic optimization lies in developing sophisticated, interpretable models that can seamlessly integrate multi-omics data to reliably predict individual patient treatment outcomes.
Single-cell and spatial transcriptomics have emerged as transformative technologies in the field of precision medicine, enabling researchers to decipher cellular heterogeneity and spatial organization within tissues at an unprecedented resolution. Unlike traditional bulk RNA sequencing, which averages gene expression across thousands of cells, single-cell RNA sequencing (scRNA-seq) captures the transcriptome of individual cells, revealing rare cell populations, transition states, and cellular dynamics that drive disease pathogenesis and treatment response [45]. The rapid advancement of scRNA-seq technologies has revolutionized our understanding of cancer biology, immune regulation, and developmental processes by providing high-resolution profiling of individual cells across genomic, transcriptomic, and epigenomic landscapes [45].
Spatial transcriptomics (ST) represents a complementary breakthrough that preserves the crucial geographical context of gene expression within intact tissue architectures. This technology integrates histological visualization with transcriptomic profiling, allowing researchers to pinpoint exactly where specific genes are active in a tissue section [46]. The preservation of spatial information is critical for understanding complex biological systems, as cellular function and behavior are fundamentally shaped by a cell's physical microenvironment and its interactions with neighboring cells [47]. The global spatial transcriptomics market, valued at $410.46 million in 2024 and projected to reach $1,569.03 million by 2034, reflects the significant impact and adoption of these technologies across research and clinical domains [46].
The integration of these technologies provides a multidimensional view of cellular states and regulatory mechanisms in health and disease. By combining single-cell resolution with spatial context, researchers can now reconstruct the intricate cellular ecosystems of human tissues, uncovering novel regulatory mechanisms and therapeutic targets that were previously obscured by analytical limitations [45]. This technological synergy is particularly powerful in precision medicine applications, where understanding patient-specific disease mechanisms at cellular and spatial resolution enables more accurate diagnostics, prognostics, and tailored therapeutic interventions.
Single-Cell RNA Sequencing Platforms
Single-cell RNA sequencing technologies have evolved substantially, with current platforms broadly categorized into full-length and tag-based methods. Full-length scRNA-seq methods, such as Smart-seq2 and Quartz-seq, sequence the entire transcript, enabling identification of transcript isoforms, alternative splicing events, and single-nucleotide polymorphisms [48]. While these methods offer comprehensive transcriptome coverage with high sequencing depth and mapping efficiency, they are limited in throughput to hundreds of cells and incur higher costs per cell with significant batch effects [48].
In contrast, tag-based scRNA-seq techniques estimate transcript abundance by sequencing the 3'-end of transcripts but achieve much higher throughput, processing tens of thousands to millions of cells. These methods incorporate cell barcodes and unique molecular identifiers (UMIs) to accurately distinguish cell types and quantify transcript copies [48]. Tag-based approaches are further subdivided into:
- Droplet-based systems (e.g., 10x Genomics Chromium) that use microfluidic devices to encapsulate individual cells with barcoded beads in nanoliter-sized water droplets [45]
- Microwell-based technologies that capture cells on plates with barcoded beads [48]
- Split-pool barcoding methods that use combinatorial indexing without physical cell separation [48]
Each approach presents distinct trade-offs in throughput, cost, and information content, requiring researchers to select platforms based on specific experimental needs and biological questions.
Spatial Transcriptomics Platforms
Spatial transcriptomics technologies can be broadly classified into two categories: imaging-based and sequencing-based approaches. Imaging-based technologies utilize single-molecule fluorescence in situ hybridization (smFISH) as their foundation, enabling highly multiplexed detection of RNA transcripts through cyclic hybridization and imaging [49]. These platforms differ primarily in their probe design, hybridization strategies, signal amplification, and gene decoding mechanisms:
- Xenium employs a hybrid approach combining in situ sequencing and hybridization, using padlock probes that undergo rolling circle amplification to enhance signal sensitivity [49]
- MERSCOPE utilizes a binary barcode strategy where each gene is assigned a unique barcode read through multiple rounds of fluorescence detection [49]
- CosMx incorporates both hybridization and optical signature approaches with an additional positional dimension for gene identification [49]
Sequencing-based technologies integrate spatially barcoded arrays with next-generation sequencing to determine transcript locations and expression levels:
- 10X Visium uses spatially barcoded RNA-binding probes attached to slides, with two workflow versions (V1 for fresh tissue and V2 for both fresh and FFPE tissue) [49]
- Visium HD employs the same technology as Visium V2 but features a significantly smaller spot size of 2μm for enhanced spatial resolution [49]
- Stereo-seq utilizes DNA nanoball (DNB) technology with oligo probes that are circularized and amplified via rolling circle amplification, achieving a center-to-center distance of just 0.5μm [49]
Table 1: Comparison of Major Spatial Transcriptomics Platforms
| Platform | Technology Type | Resolution | Genes Detected | Tissue Compatibility | Key Applications |
|---|---|---|---|---|---|
| 10X Visium | Sequencing-based | 55μm spots | Whole transcriptome (limited by capture) | Fresh frozen & FFPE (with V2) | Tumor microenvironment, developmental biology |
| Visium HD | Sequencing-based | 2μm bins | Whole transcriptome | Fresh frozen & FFPE | Cellular and subcellular spatial mapping |
| Xenium | Imaging-based | Single-cell | 100s-1000s (panel-based) | FFPE & fresh frozen | High-plex targeted spatial analysis |
| MERSCOPE | Imaging-based | Single-cell | 100s-1000s (panel-based) | FFPE & fresh frozen | Cell typing, cell-cell interactions |
| CosMx | Imaging-based | Single-cell | 1000-6000 (panel-based) | FFPE & fresh frozen | High-plex targeted spatial analysis |
| Stereo-seq | Sequencing-based | 0.5μm (DNB center) | Whole transcriptome | Fresh frozen & FFPE | Large tissue areas, high-resolution mapping |
Performance Benchmarking
Recent systematic comparisons of spatial transcriptomics platforms provide critical insights for technology selection. A comprehensive evaluation of sequencing-based ST methods across reference tissues revealed significant variability in molecular diffusion, which substantially affects effective resolution [47]. When comparing molecule-capture efficiency across platforms, Stereo-seq demonstrated the highest capturing capability with regular array sizes up to 1cm, while Slide-seq V2 was limited to partial tissue coverage due to constrained capture size [47].
In a rigorous assessment of imaging-based ST platforms using formalin-fixed paraffin-embedded (FFPE) tumor samples, CosMx detected the highest transcript counts and uniquely expressed gene counts per cell, though it exhibited challenges with target gene probes expressing at levels similar to negative controls in some samples [50]. Xenium with unimodal segmentation showed higher transcript and gene counts per cell than its multimodal counterpart, with minimal target genes expressing similarly to negative controls [50]. These performance characteristics have direct implications for data quality and interpretation in precision oncology applications.
Table 2: Performance Metrics from Spatial Transcriptomics Benchmarking Studies
| Platform | Transcripts/Cell | Unique Genes/Cell | Sensitivity | Concordance with RNA-seq | Cell Segmentation Accuracy |
|---|---|---|---|---|---|
| CosMx | Highest among tested platforms | Highest among tested platforms | Variable across tissue types | Moderate to high | Manufacturer algorithm performed well |
| Xenium (UM) | Moderate to high | Moderate to high | High | High | Good nuclear segmentation |
| Xenium (MM) | Lower than UM | Lower than UM | High | High | Improved cytoplasm coverage |
| MERSCOPE | Moderate | Moderate | Higher in newer tissues | Moderate | Affected by tissue morphology |
| Visium (probe-based) | High in downsampled data | High in downsampled data | High in specific regions | High | Limited by spot size (55μm) |
| Stereo-seq | High with full sequencing | High with full sequencing | Highest with full data | High | Limited by binning strategy |
Experimental Design and Methodologies
Sample Preparation Protocols
Obtaining high-quality single-cell suspensions or properly preserved tissue sections is a critical determinant of success in single-cell and spatial transcriptomics studies. For single-cell sequencing, the process begins with tissue dissociation through mechanical disruption and enzymatic digestion, preferably using automatic tissue dissociators to minimize batch effects [48]. Optimization of dissociation protocols is essential, as excessive dissociation causes cell damage, reduces viability, and introduces unwanted transcriptional changes, while insufficient dissociation leads to multiplets in the data [48]. Cell viability and integrity must be carefully monitored, with filtration through appropriately sized cell strainers or debris removal solutions to ensure clean suspensions.
For spatial transcriptomics, tissue preservation and sectioning are crucial steps. Optimal protocols depend on the technology platform and sample type. For sequencing-based approaches like 10X Visium, tissues are typically flash-frozen in optimal cutting temperature (OCT) compound or fixed in formalin and embedded in paraffin (FFPE) [49]. Section thickness varies by platform, with 5-10μm being common for most applications. Proper tissue collection, preservation, and sectioning maintain RNA integrity while preserving spatial context, enabling accurate transcript localization and quantification.
Recent advancements address traditional limitations through single-nucleus RNA sequencing (snRNA-seq), which enables analysis of frozen tissues and tissues difficult to dissociate, such as neuronal samples [48]. While snRNA-seq reduces cell stress and composition bias, it provides information primarily on nuclear RNA and includes various intron sequences, requiring careful consideration of experimental objectives when selecting between single-cell and single-nucleus approaches.
Quality Control and Normalization
Rigorous quality control is essential for generating reliable single-cell and spatial data. For scRNA-seq, standard quality metrics include the number of UMIs per cell, genes detected per cell, and the proportion of mitochondrial genes [48]. Cells with low UMI counts, few detected genes, or high mitochondrial content typically indicate poor viability or compromised cell integrity and should be filtered out. For spatial transcriptomics, additional quality measures include assessment of tissue morphology, RNA retention, and background signal using negative control probes [50].
Normalization addresses technical variations in sequencing depth, capture efficiency, and other platform-specific artifacts. For scRNA-seq data, scaling gene expression counts by the total number of sequencing reads or counts per cell is standard practice [48]. More advanced normalization methods account for batch effects, which can arise from different protocols, sample handling, and platforms [48]. For spatial data, normalization must consider spatial artifacts, uneven tissue permeabilization, and background noise, often requiring specialized computational approaches tailored to specific technologies.
Analytical Workflows
The analytical pipeline for single-cell and spatial transcriptomics involves multiple computational steps:
Data Preprocessing: Raw sequencing data are processed using specialized pipelines (e.g., Cell Ranger for 10X Genomics data) to generate gene-by-cell count matrices [48]
Feature Selection and Dimensionality Reduction: Identification of highly variable genes followed by principal component analysis (PCA) to reduce dimensionality [48]
Clustering and Cell Type Annotation: Unsupervised clustering algorithms (e.g., in Seurat package) group cells based on gene expression patterns, followed by annotation using marker genes [48]
Spatial Data Integration: For spatial transcriptomics, expression data are mapped to spatial coordinates, enabling analysis of spatial patterns, neighborhood relationships, and cell-cell communication [47]
Trajectory Inference and Differential Expression: Pseudotime analysis reconstructs developmental trajectories, while differential expression testing identifies genes associated with specific conditions, cell types, or spatial regions [45]
The following workflow diagram illustrates the key experimental and computational steps in a comprehensive single-cell and spatial transcriptomics study:
Diagram 1: Single-Cell and Spatial Transcriptomics Workflow. The process begins with tissue collection and proceeds through sample preparation, library construction, sequencing/imaging, and computational analysis to biological interpretation.
Applications in Precision Medicine
Oncology and Cancer Heterogeneity
Single-cell and spatial transcriptomics have revolutionized our understanding of cancer biology by elucidating intratumoral heterogeneity, clonal evolution, and the complex ecosystem of the tumor microenvironment (TME). Single-cell DNA sequencing (scDNA-seq) has proven particularly valuable for interrogating intratumoral heterogeneity and clonal evolution across numerous malignancies [45]. This approach enables researchers to reconstruct the evolutionary trajectories of tumor subclones, revealing rare populations such as cancer stem cells or therapy-resistant clones that drive disease progression and treatment failure [45].
The application of these technologies in translational oncology is exemplified by studies of circulating tumor cells (CTCs). Research by Polzer et al. utilized scDNA-seq to analyze CTC genomic profiles in breast cancer patients, identifying fundamental principles of tumor evolution, including the generation of genetic diversity through mutations and chromosomal rearrangements, and clonal selection of subpopulations with metastatic potential or therapy resistance [45]. These findings provide new perspectives on CTC dynamics, highlighting the coexistence of genetically distinct subpopulations with unique therapeutic vulnerabilities.
In solid tumors, spatial transcriptomics has uncovered the organizational principles of the TME and its role in therapeutic response. A comprehensive comparison of imaging-based ST platforms using FFPE tumor samples demonstrated the ability to identify distinct cellular neighborhoods and their association with clinical outcomes [50]. These spatial patterns provide critical insights for immuno-oncology, enabling researchers to understand why some patients respond to immunotherapy while others do not, based on the spatial arrangement of immune and tumor cells.
Predictive Modeling and Therapeutic Discovery
The integration of single-cell data with machine learning approaches has opened new avenues for predictive modeling and therapeutic discovery in precision medicine. The scTherapy platform represents a groundbreaking application of this integration, leveraging single-cell transcriptomic profiles to prioritize multi-targeting treatment options for individual cancer patients [51]. This machine learning approach addresses the critical challenge of intratumoral heterogeneity by predicting drug combinations that selectively co-inhibit multiple cancer subclones while minimizing toxicity to normal cells.
In experimental validations using primary cells from acute myeloid leukemia (AML) patients, scTherapy demonstrated remarkable performance, with 96% of predicted multi-targeting treatments exhibiting selective efficacy or synergy, and 83% showing low toxicity to normal cells [51]. This approach enables systematic tailoring of personalized combination therapies by considering both cellular heterogeneity and dose-specific therapeutic effects, significantly expanding the combinatorial space that can be tested in scarce patient-derived cells.
The following diagram illustrates the scTherapy computational framework for predicting patient-specific combination therapies:
Diagram 2: scTherapy Prediction Framework. The computational pipeline processes patient-specific single-cell RNA-seq data to identify cancer subclones, extracts differentially expressed genes (DEGs), and applies a pre-trained machine learning model to predict effective multi-targeting therapies.
Clinical Translation and Biomarker Discovery
The translation of single-cell and spatial technologies into clinical practice is advancing rapidly across medical specialties. In nephrology, single-cell analysis has identified disease-related biomarkers and pathways in conditions such as chronic kidney disease (CKD) and acute kidney injury (AKI), enabling more accurate patient classification and tailored treatments [48]. Similar applications are emerging in neurodegenerative diseases, cardiovascular disorders, and autoimmune conditions, where cellular heterogeneity plays a crucial role in disease pathogenesis.
Spatial transcriptomics has proven particularly valuable for companion diagnostic development in oncology. A comparative study demonstrated that spatial phenotyping had the highest predictive value for immunotherapy success when compared to next-generation sequencing, RNA expression, and standard immunohistochemistry testing [52]. This enhanced predictive capability stems from the ability to assess not just the presence of biomarkers, but their spatial organization and cellular context within the tumor microenvironment.
The integration of multiomics data layers further enhances clinical applications. Proteogenomics—which combines proteomic, transcriptomic, and genomic data—has been applied to ovarian cancer, revealing how genomic alterations drive proteomic changes and identifying factors associated with treatment outcomes and survival [52]. These integrated approaches provide a more comprehensive understanding of disease mechanisms and facilitate the development of more accurate diagnostic, prognostic, and predictive biomarkers.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful single-cell and spatial transcriptomics studies require careful selection of reagents and materials optimized for specific technologies and sample types. The following table details essential components of the experimental toolkit:
Table 3: Essential Research Reagents and Solutions for Single-Cell and Spatial Transcriptomics
| Reagent Category | Specific Examples | Function | Technical Considerations |
|---|---|---|---|
| Tissue Preservation Reagents | OCT compound, RNAlater, Formalin, Paraffin | Maintain tissue architecture and RNA integrity | Choice depends on technology: frozen for most scRNA-seq, FFPE for many spatial platforms |
| Dissociation Kits | Tumor Dissociation Kits, Neural Tissue Dissociation Kits | Liberate individual cells from tissue matrix | Must be optimized for specific tissue types to balance yield and viability |
| Viability Stains | Trypan Blue, Propidium Iodide, DAPI, Calcein AM | Assess cell integrity and exclude dead cells | Critical for scRNA-seq; dead cells increase ambient RNA |
| Capture Beads/Oligos | 10x Barcoded Beads, Stereo-seq DNBs | Bind mRNA and incorporate spatial/cellular barcodes | Platform-specific; determine cellular throughput and capture efficiency |
| Library Preparation Kits | Chromium Next GEM Kits, SMART-seq HT Kits | Convert RNA to sequencing-ready libraries | Determine sequencing compatibility, sensitivity, and bias |
| Enzymatic Mixes | Reverse Transcriptase, Amplification Polymerases | cDNA synthesis and amplification | Affect fidelity, bias, and success with degraded samples |
| Probe Panels | CosMx Human Universal Panel, Xenium Gene Panels | Target-specific gene detection in spatial platforms | Determine gene coverage and application focus (e.g., immuno-oncology) |
| Imaging Reagents | Fluorophore-conjugated antibodies, Dyes | Visualize tissue morphology and protein markers | Enable multimodal integration of transcriptomic and proteomic data |
| Bioinformatics Tools | Cell Ranger, Seurat, Space Ranger | Process raw data and perform quality control | Essential for data interpretation; require computational expertise |
Integration with Multi-Omics and Artificial Intelligence
The true power of single-cell and spatial technologies emerges when integrated with other data modalities and advanced computational approaches. Multi-omics integration combines genomic, transcriptomic, epigenomic, proteomic, and metabolomic data to provide a comprehensive understanding of cellular states and regulatory mechanisms [53]. This integrated approach is particularly powerful in precision medicine, where it helps decipher the complex interplay between genetic predisposition, environmental factors, and disease manifestations.
Artificial intelligence (AI) and machine learning (ML) algorithms are transforming the analysis of single-cell and spatial data by enabling more efficient processing of large-scale datasets, identification of complex patterns, and enhanced prediction of gene expression and cellular behavior [46]. AI approaches manage the computational demands of multidimensional data, detect subtle patterns and biomarkers that might be overlooked by traditional methods, and facilitate integration across different technologies and experimental conditions [46]. These capabilities are driving market expansion and accelerating discoveries across diverse research domains.
The integration of single-cell data with electronic health records (EHRs) represents another frontier in precision medicine. By combining high-resolution molecular profiles with clinical data, researchers can establish more robust associations between cellular features and patient outcomes, enabling more accurate risk stratification and treatment selection [53]. This approach is particularly valuable for pediatric health care, where understanding the developmental context of disease can inform early intervention strategies.
Single-cell and spatial transcriptomics technologies have fundamentally transformed biomedical research and are increasingly influencing clinical practice in precision medicine. These approaches provide unprecedented insights into cellular heterogeneity, tissue organization, and disease mechanisms, enabling more accurate diagnosis, prognosis, and therapeutic targeting. The continuous evolution of these technologies—with improvements in resolution, sensitivity, throughput, and accessibility—promises to further enhance their impact in the coming years.
The future trajectory of these fields will likely focus on several key areas: (1) enhanced multi-omics integration at the single-cell level, combining genomic, epigenomic, transcriptomic, proteomic, and metabolomic measurements from the same cells; (2) development of more sophisticated computational tools, particularly AI and ML approaches, to extract biological insights from increasingly complex datasets; (3) standardization of protocols and analytical frameworks to improve reproducibility and clinical translation; and (4) expansion of diverse population representation in genomic and transcriptomic databases to ensure equitable benefits from precision medicine advances [53].
As these technologies continue to mature and integrate with other data modalities, they will increasingly enable a comprehensive, high-resolution view of human health and disease. This paradigm shift from population-level averages to individual cellular profiling represents the foundation for truly personalized medical approaches that consider each patient's unique disease biology, ultimately leading to more effective interventions and improved clinical outcomes across diverse conditions and populations.
Overcoming Challenges and Enhancing Model Performance
Addressing Data Redundancy and Collinearity Between Omics Layers
The integration of multiple omics layers into genomic prediction models represents a significant frontier in agricultural genetics and biomedical research. While genomic selection has traditionally relied on DNA-based markers to predict breeding values, attention has now turned to incorporating additional molecular data layers, particularly transcriptomics, to improve prediction accuracy. However, this integration introduces substantial statistical challenges, primarily stemming from the inherent biological relationships between these data types. Because gene expression is itself partially heritable, transcriptomic data captures both genetic and environmental influences, creating natural collinearity between genomic and transcriptomic information. When both SNP genotypes and transcriptomic data are used as independent random effects in mixed linear models, this redundancy leads to model instability and variance component estimation biases. Addressing this collinearity is therefore paramount for developing robust multi-omics prediction models that accurately dissect the contributions of each biological layer to complex traits.
Statistical Methods for Addressing Omics Redundancy
Conceptual Approaches to Data Integration
Researchers have developed several statistical frameworks to handle the redundancy between omics layers. These approaches generally fall into two categories: those that explicitly partition the variance components and those that condition one omics layer on another. The Christensen et al. method employs a two-step procedure that first estimates the total effect of omics data on phenotypes and then explicitly models the genetic portion of these omics effects in a second step [16] [3]. In contrast, the Perez et al. method uses a one-step approach that models genotype data and omics data conditioned on the genotypes simultaneously, ensuring that the modeled omics effects are purely non-genetic [16] [3]. A derived GTCBLUPi model builds upon this concept by specifically addressing redundant information between genomic and transcriptomic information, creating conditioned transcriptomic effects that capture only the unique information not explained by genetics [16] [28] [3].
Model Specifications and Mathematical Formulations
The statistical models for handling omics redundancy share a common foundation in mixed model methodology but differ in their treatment of variance components. The standard GBLUP model serves as a baseline, specified as: y = Xb + Zgg + e, where y is the vector of phenotypes, X is the incidence matrix for fixed effects, g represents random additive genetic effects based on genomic relationships, and e is the residual term [16] [3]. The TBLUP model adapts this framework for transcriptomic data: y = Xb + Ztt + e, replacing genomic effects with transcriptomic effects (t) based on transcript abundance similarity matrices [16] [3]. The naive GTBLUP model simply combines both effects: y = Xb + Zgg + Ztt + e, but suffers from collinearity issues [3]. The advanced GTCBLUPi model addresses this limitation: y = Xb + Zgg + Zctc + e, where tc represents transcriptomic effects conditioned on genetic effects to remove shared variation [3]. This conditioning is mathematically achieved by deriving the transcriptomic relationship matrix conditional on the genomic relationship matrix, effectively orthogonalizing the two random effects.
Quantitative Performance Comparison Across Models
Variance Component Explanations
Table 1: Proportion of Phenotypic Variance Explained by Different Omics Layers in Japanese Quail Efficiency Traits
| Trait | Genomic Variance (GBLUP) | Transcriptomic Variance (TBLUP) | Combined Variance (GTBLUP) | Conditioned Variance (GTCBLUPi) |
|---|---|---|---|---|
| Phosphorus Utilization (PU) | 0.21 | 0.45 | 0.52 | 0.49 |
| Body Weight Gain (BWG) | 0.18 | 0.39 | 0.44 | 0.42 |
| Feed Conversion Ratio (FCR) | 0.16 | 0.42 | 0.47 | 0.45 |
| Tibia Ash (TA) | 0.23 | 0.48 | 0.55 | 0.52 |
| Calcium Utilization (CaU) | 0.19 | 0.41 | 0.46 | 0.44 |
Data adapted from the Japanese quail study on efficiency traits [16] [3]. The transcriptomic data consistently explained a larger portion of phenotypic variance than genomic data alone across all traits. Models incorporating both genetic and transcriptomic information (GTBLUP) outperformed single-omics models, but the conditioned approach (GTCBLUPi) provided more stable variance partitioning by addressing collinearity.
Prediction Accuracy Comparisons
Table 2: Prediction Accuracy (Pearson Correlation) for Efficiency Traits Using Different Models
| Trait | GBLUP | TBLUP | GTBLUP | GTCBLUPi |
|---|---|---|---|---|
| Phosphorus Utilization (PU) | 0.47 | 0.62 | 0.68 | 0.71 |
| Body Weight Gain (BWG) | 0.43 | 0.58 | 0.63 | 0.66 |
| Feed Conversion Ratio (FCR) | 0.41 | 0.59 | 0.65 | 0.67 |
| Tibia Ash (TA) | 0.49 | 0.64 | 0.70 | 0.72 |
| Calcium Utilization (CaU) | 0.44 | 0.60 | 0.66 | 0.68 |
The GTCBLUPi model, which specifically addresses redundancy between omics layers, achieved the highest prediction accuracy for all efficiency traits in the Japanese quail study [16] [3]. The improvement over naive combination models (GTBLUP) demonstrates the value of properly handling collinearity, particularly for complex traits influenced by both genetic and regulatory mechanisms.
Experimental Protocols for Multi-Omics Integration
Japanese Quail Study Design
The primary dataset for evaluating omics redundancy came from a comprehensive study of 480 F2 cross Japanese quails raised under controlled conditions [16] [3]. The experimental population was derived from mating 12 males and 12 females from each founder line to produce the F1 generation, followed by random selection of 17 roosters and 34 hens from the F1 to produce the F2 generation. All birds were fed an ad libitum corn-soybean meal-based diet with marginal phosphorus concentration to elicit genetic potential for phosphorus utilization. Phenotypic measurements included phosphorus utilization, body weight gain, feed intake, feed conversion ratio, tibia ash, and calcium utilization. Blood and ileum mucosa samples were collected at day 15 for genotyping and transcriptomic analysis [16] [3].
Omics Data Generation and Processing
Genotyping was performed using a 6k Illumina iSelect chip, filtered to 4k high-quality SNPs for analysis [16] [3]. For transcriptomic profiling, ileal microRNA and messenger RNA sequencing was conducted on discordant sib pairs selected from ten families, with one sib exhibiting high and the other low phosphorus utilization. Differential expression analysis identified 77 miRNAs and 80 mRNAs associated with phosphorus utilization, which were then assessed using 96.96 dynamic arrays on a Fluidigm BioMark HD system for the entire subpopulation of 480 quails [16] [3]. Phenotypic data transformation was applied using Box-Cox transformation with trait-specific lambda parameters to address distribution skewness before model fitting.
Statistical Analysis Workflow
The analytical workflow proceeded through several stages: (1) quality control and normalization of omics data; (2) calculation of genomic and transcriptomic relationship matrices; (3) fitting of separate GBLUP and TBLUP models to establish baseline performance; (4) implementation of combined models (GTBLUP) without redundancy adjustment; and (5) application of conditioned models (GTCBLUPi) to address collinearity. Variance components were estimated using restricted maximum likelihood (REML) in ASReml-R software, and prediction accuracies were evaluated through cross-validation procedures [16] [3].
Diagram 1: Experimental workflow for multi-omics model development and evaluation, highlighting the key steps from data generation through conditioned model implementation.
Table 3: Essential Research Tools and Reagents for Multi-Omics Prediction Studies
| Tool/Resource | Category | Specific Function | Example Implementation |
|---|---|---|---|
| ASReml-R | Statistical Software | Fitting mixed linear models with variance component estimation | REML estimation for GBLUP, TBLUP, and GTCBLUPi models [16] [3] |
| Fluidigm BioMark HD | Laboratory Instrument | High-throughput transcriptomic profiling | Dynamic array-based quantification of miRNA and mRNA candidates [16] [3] |
| Illumina iSelect | Genotyping Platform | Genome-wide SNP genotyping | 6k SNP array for genomic relationship matrix calculation [16] [3] |
| glmnet R Package | Statistical Tool | Penalized regression for high-dimensional data | Ridge regression and LASSO implementation for transcriptomic prediction [54] |
| PLS R Package | Statistical Tool | Dimension reduction for omics data | Partial least squares regression for gene expression data [54] |
| AlphaSimR | Simulation Tool | Genomic breeding simulation | Generating synthetic populations with defined genetic architectures [35] |
| EasyGeSe | Benchmarking Resource | Standardized genomic prediction evaluation | Multi-species dataset collection for method comparison [7] |
Biological Insights from Multi-Omics Integration
Beyond statistical improvements, properly handling omics redundancy enables deeper biological insights into trait architecture. The Japanese quail study revealed that transcript abundances from intestinal tissue explained a larger portion of phenotypic variance for efficiency traits than host genetics alone [16] [3]. This suggests that transcriptional regulation in metabolic tissues captures substantial environmental influences and gene-environment interactions that are not encoded in the DNA sequence. Additionally, the study identified high transcriptomic correlations between efficiency traits, indicating shared regulatory pathways that might be targeted for simultaneous improvement of multiple traits. The biological interpretation of these relationships is only possible when variance components are properly partitioned using conditioned models like GTCBLUPi, as naive combinations would inflate the apparent contribution of genomics due to shared variance with transcriptomics.
Addressing redundancy and collinearity between omics layers is not merely a statistical refinement but a fundamental requirement for accurate biological interpretation and prediction. The GTCBLUPi framework and related conditioning approaches provide a robust solution that acknowledges the biological relationships between molecular layers while enabling precise dissection of their unique contributions to complex traits. As multi-omics data becomes increasingly accessible in both agricultural and biomedical contexts, these methods will play a crucial role in maximizing the predictive value of integrated molecular data. Future methodological development should focus on extending these principles to additional omics layers, including metabolomics and proteomics, and developing computationally efficient implementations suitable for large-scale breeding and biomedical applications.
The integration of genomic and transcriptomic data into prediction models represents a transformative advancement in biomedical and agricultural research. However, the generation of high-quality, large-scale RNA sequencing (RNA-Seq) data remains cost-prohibitive for many research programs, particularly those requiring substantial sample sizes for robust statistical power. The fundamental challenge lies in balancing data quality and coverage with financial constraints, as traditional RNA-Seq protocols consume significant reagents and sequencing resources. This comprehensive guide examines two complementary strategic approaches—experimental miniaturization and sequencing depth optimization—that enable researchers to substantially reduce costs while maintaining data integrity for genomic prediction models. By implementing these methodologies, research programs can enhance their scalability and accelerate discoveries in genomic medicine and agricultural genomics.
Recent advances in genomic selection have demonstrated that transcriptomic data often explains a larger portion of phenotypic variance than genomic data alone, highlighting the critical value of RNA-Seq information for predicting complex traits [16]. However, without cost-effective strategies, the generation of such data remains inaccessible for many large-scale studies. The protocols and data comparisons presented herein provide a roadmap for researchers to maximize their resource utilization while generating high-quality data for both genomic and transcriptomic prediction models.
Experimental Miniaturization: Scaling Down to Scale Up
Protocol Fundamentals and Workflow Optimization
Experimental miniaturization refers to the systematic reduction of reagent volumes and the integration of automation to process samples at higher throughput with lower per-sample costs. This approach has been successfully demonstrated in plant genomics research, where a miniaturized RNA extraction protocol reduced reagent volumes by 50% while maintaining RNA quality standards [55]. Similarly, a miniaturized library preparation method utilizing the VAHTS Universal V6 RNA-seq Library Prep Kit successfully scaled down reagent volumes to just 25% of the manufacturer's original recommendations without compromising library quality or complexity [55].
The critical success factors for experimental miniaturization include:
- Liquid Handling Automation: Precision instrumentation such as the Mosquito HV and Dragonfly Discovery platforms enables accurate nanoliter-volume liquid transfers, eliminating the manual pipetting errors that often plague miniaturized protocols [56].
- Workflow Integration: Successful miniaturization requires optimizing the entire experimental workflow from RNA extraction through library preparation rather than focusing on individual steps in isolation.
- Quality Control Checkpoints: Implementing rigorous QC at each stage (RNA quality, cDNA synthesis, final library) ensures that volume reduction does not introduce technical variability or bias.
Table 1: Miniaturized RNA-Seq Workflow Components and Specifications
| Protocol Step | Traditional Volume | Miniaturized Volume | Key Adaptations |
|---|---|---|---|
| RNA Extraction | 100μL TRIzol per sample | 50μL TRIzol per sample | Additional ethanol wash to remove residual phenol [55] |
| mRNA Selection | Full volume poly-A capture | 25% volume poly-A capture | Reduced binding buffer and wash volumes [55] |
| Library Preparation | Manufacturer's recommended volume | 25% of recommended volume | Automated liquid handling with precision instruments [56] |
| PCR Amplification | 25-50μL reactions | 10-17.5μL reactions | Optimized cycle numbers to maintain complexity [56] |
Implementation and Practical Considerations
The transition to miniaturized protocols requires careful validation specific to each laboratory's research context. Researchers should conduct pilot studies comparing miniaturized versus standard protocols using representative sample types to verify that data quality remains consistent. Special attention should be paid to potential batch effects introduced by automated platforms, though studies have demonstrated that with proper calibration, automated liquid handling introduces less technical variability than manual processing [56].
For laboratories processing hundreds to thousands of samples, the integration of combinatorial indexing strategies can further enhance throughput while controlling costs. These methods utilize multi-step barcoding approaches that dramatically increase the number of samples that can be multiplexed in a single sequencing run [57]. While initially developed for single-cell RNA-Seq, the principles of combinatorial indexing can be adapted to bulk RNA-Seq applications to maximize sample throughput.
Sequencing Depth Reduction: Maximizing Information per Read
Strategic Depth Optimization Approaches
Sequencing depth reduction strategies focus on allocating sequencing resources more efficiently to minimize redundant coverage while maintaining statistical power for variant detection and expression quantification. The SPRE-Seq (Specific-Regions-Enriched sequencing) methodology represents an innovative approach that enables differential depth sequencing within a single assay [58]. This technique uses streptavidin pre-blocking of oligonucleotide probes to strategically enrich for specific genomic regions of interest, thereby allocating greater sequencing depth to regions with higher biological significance or technical challenges.
In practice, SPRE-Seq applied to a custom homologous recombination deficiency (HRD) assay demonstrated that the effective sequencing depths of target regions met required thresholds with only half the sequencing data volume (reduced from 12 to 6 GB) while maintaining 100% consistency with expected results for HRR genes and HRD status [58]. This approach recognizes that uniform high-depth sequencing across all genomic regions is inefficient, as different regions have varying depth requirements for accurate variant calling.
Table 2: Sequencing Depth Reduction Strategies and Performance Metrics
| Strategy | Methodology | Data Volume Reduction | Performance Maintenance |
|---|---|---|---|
| SPRE-Seq | Streptavidin pre-blocked probes for differential enrichment | 50% reduction (12GB to 6GB) | 100% consistency for target regions [58] |
| WEGS | Combined low-depth WGS (2-5X) with high-depth WES (100X) | 1.7-2.0x cheaper than standard WES | Similar precision/recall for coding variants [59] |
| Multiplexed Sequencing | Sample pooling (4-8 plex) with unique barcodes | 1.7-2.3x increase in duplicated reads | Maintained variant calling accuracy with UMI correction [59] |
Multiplexing and Computational Optimization
Sample multiplexing, where multiple samples are pooled and sequenced simultaneously using unique barcodes, represents another powerful strategy for reducing per-sample sequencing costs. However, this approach introduces technical challenges, including increased rates of PCR duplicates—with 4-plex and 8-plex experiments showing 1.7-fold and 2.3-fold increases in duplicated reads respectively [59]. The implementation of unique molecular identifiers (UMIs) helps mitigate this issue by distinguishing truly duplicated fragments from PCR artifacts, though the effectiveness varies between computational tools [59].
The emerging consensus suggests that rather than applying uniform depth across all samples, researchers should implement tiered depth strategies based on sample priority and research questions. For instance, key discovery cohorts might be sequenced at higher depth while validation cohorts utilize lower depth, supplemented by imputation from high-depth reference sets.
Performance Comparison: Genomic vs. Transcriptomic Prediction Models
Predictive Accuracy in Complex Traits
Direct comparisons between genomic and transcriptomic prediction models reveal distinct advantages for each approach depending on the biological context and trait architecture. In a comprehensive study of efficiency-related traits in Japanese quail, models incorporating transcriptomic data derived from intestinal tissue explained a larger portion of phenotypic variance than models based solely on host genetics [16]. The integration of both data types consistently produced the highest prediction accuracies, demonstrating that transcriptomic information effectively complements rather than replaces genomic data.
Similarly, in plant genomics, RNA-Seq data for barley recombinant inbred lines achieved genomic prediction performance comparable to or better than traditional SNP array datasets when combined with parental whole-genome sequencing SNP data [55]. This hybrid approach demonstrated particular strength in inter-population predictions, where training and validation sets originated from different sub-populations, suggesting that transcriptomic data may capture environmental influences and gene regulation patterns that improve generalizability across diverse populations.
Table 3: Performance Comparison of Prediction Models for Agricultural Traits
| Model Type | Data Sources | Prediction Ability | Optimal Application Context |
|---|---|---|---|
| GBLUP | Genomic markers only | 0.73-0.78 (barley study) | Baseline for genetic value prediction [55] |
| TBLUP | Transcriptomic data only | Higher than GBLUP for some traits | When gene expression captures environmental responses [16] |
| GTCBLUPi | Integrated genomic & transcriptomic | Highest overall accuracy | Complex traits with gene-environment interactions [16] |
Cost-Benefit Analysis for Research Planning
When evaluating the cost-effectiveness of different data generation strategies, researchers must consider both experimental costs and predictive performance. The WEGS (Whole Exome Genome Sequencing) approach, which combines low-depth whole genome sequencing (2-5X) with high-depth whole exome sequencing (100X), provides a balanced solution at 1.7-2.0 times cheaper than standard WES and 1.8-2.1 times cheaper than 30X WGS [59]. This method maintains similar precision and recall rates for rare coding variants while capturing population-specific variants in non-coding regions that are difficult to recover through imputation.
For transcriptomic prediction, the miniaturization strategies described in Section 2 can reduce per-sample costs by approximately 60-70% through reagent volume reduction and process automation [55]. When combined with optimized sequencing depth, these approaches make large-scale transcriptomic profiling feasible for breeding programs and biomedical studies with limited resources.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful implementation of cost-effective RNA-Seq strategies requires specific reagents and platforms optimized for miniaturization and high-throughput processing. The following solutions have been experimentally validated in the studies cited throughout this guide:
- TRIzol Reagent: Adapted to 50% volume in 96-well format for high-throughput RNA extraction while maintaining RNA integrity [55].
- VAHTS Universal V6 RNA-seq Library Prep Kit: Successfully miniaturized to 25% reaction volumes without compromising library complexity or representation [55].
- Illumina COVIDSeq Assay: Demonstrated compatibility with automated miniaturization protocols for large-scale studies [56].
- Mosquito HV and Dragonfly Discovery Platforms: Precision liquid handling instruments capable of accurate nanoliter-volume transfers essential for miniaturized protocols [56].
- Twist Fast Hybridization and Wash Kit: Used in capture-based targeted sequencing approaches like SPRE-Seq for differential depth sequencing [58].
- Unique Molecular Identifiers (UMIs): Critical for mitigating PCR duplication artifacts in multiplexed sequencing designs, with implementation tools like LocatIt and GATK+UMI offering different trade-offs in depth preservation versus quality control [59].
Integrated Workflow and Decision Framework
The following diagram illustrates the key decision points and methodological options for implementing cost-effective RNA-Seq strategies:
The strategic implementation of RNA-Seq miniaturization and sequencing depth reduction enables researchers to overcome the economic barriers that often limit sample size and statistical power in genomic studies. By carefully selecting and validating appropriate cost-reduction strategies based on specific research objectives, scientific teams can generate high-quality data for both genomic and transcriptomic prediction models at significantly reduced costs. The experimental evidence presented in this guide demonstrates that these approaches maintain data integrity while expanding research capabilities, particularly for large-scale studies in both biomedical and agricultural contexts. As genomic prediction models continue to evolve, these cost-effective data generation strategies will play an increasingly vital role in accelerating scientific discovery and practical applications across diverse research domains.
In the field of genomic selection, the integration of transcriptomic data with traditional genomic markers presents a powerful approach to enhance phenotypic prediction accuracy. However, this integration introduces significant technical challenges related to data heterogeneity, standardization, and workflow management. As researchers increasingly recognize that transcript abundances can explain a larger portion of phenotypic variance than host genetics alone for certain traits, the development of robust computational frameworks becomes paramount [3] [16]. The inherent redundancy between genomic and transcriptomic information layers, coupled with their different dimensionalities, measurement scales, and biological interpretations, demands sophisticated statistical models and reproducible bioinformatics workflows [3] [17]. This guide examines the current landscape of multi-omics prediction models, comparing their performance across species and experimental designs, while providing practical solutions for managing the technical complexities of heterogeneous data integration.
Performance Comparison: Genomic vs. Transcriptomic Prediction Models
Quantitative Performance Metrics Across Studies
Table 1: Comparison of prediction accuracy between different omics models across multiple studies
| Study Organism | GBLUP Accuracy (Genomic) | TBLUP Accuracy (Transcriptomic) | Combined Model Accuracy | Top Performing Model | Key Findings |
|---|---|---|---|---|---|
| Japanese Quail (n=480) [3] | Moderate | Higher than GBLUP | Highest | GTCBLUPi | Transcript abundances explained larger phenotypic variance than genetics alone |
| Barley RIL Populations [5] | 0.73-0.78 (SNP array) | Comparable to SNP | 0.73-0.78 | Consensus SNP (RNA-Seq + WGS) | RNA-Seq data achieved prediction ability comparable to traditional SNP arrays |
| Maize282 [17] | Variable by trait | Variable by trait | Consistently improved | Model-based fusion | Model-based fusion outperformed simple concatenation approaches |
Interpretation of Comparative Results
The consistent pattern across multiple studies indicates that integrated models leveraging both genomic and transcriptomic data generally outperform single-omics approaches. The Japanese quail study demonstrated that transcriptomic data from ileum tissue explained a larger portion of phenotypic variance for efficiency-related traits than genomic data alone [3]. Similarly, in barley, RNA-Seq data achieved genomic prediction performance comparable to or better than traditional SNP array datasets, with the consensus SNP dataset (combining RNA-Seq and parental whole-genome sequencing) performing best, particularly in inter-population predictions where training and validation sets originated from different recombinant inbred line sub-populations [5].
The performance advantage of multi-omics integration is especially pronounced for complex traits influenced by multiple biological pathways. Studies across species reveal that the choice of integration strategy significantly impacts success, with model-based fusion techniques that capture non-additive, nonlinear, and hierarchical interactions consistently outperforming simple data concatenation approaches [17].
Experimental Protocols and Methodologies
Standardized Model Architectures for Multi-Omics Integration
Table 2: Statistical models for multi-omics data integration
| Model Acronym | Full Name | Description | Key Features | Implementation Considerations |
|---|---|---|---|---|
| GBLUP [3] | Genomic Best Linear Unbiased Prediction | Uses genome-wide SNPs to predict breeding values | Industry standard for genomic selection | Sensitive to population structure; limited for complex traits |
| TBLUP [3] | Transcriptomic Best Linear Unbiased Prediction | Uses transcript abundances to predict phenotypes | Captures functional activity; tissue-specific | Conditionally dependent on genetics and environment |
| GTBLUP [3] | Genomic-Transcriptomic BLUP | Combines both SNP and transcriptomic data as independent effects | Simple integration approach | Susceptible to collinearity between data layers |
| GTCBLUPi [3] | Genomic-Transcriptomic Conditional BLUP | Models transcriptomic data conditioned on genotypes | Addresses redundancy between omics layers; one-step approach | More complex implementation but superior performance |
Data Processing Workflows
The experimental protocols for multi-omics prediction share common elements despite species differences. The Japanese quail study employed Box-Cox transformation with trait-specific lambda parameters to address highly skewed phenotypic distributions, followed by scaling and centering of transformed phenotypes [3]. Genomic relationship matrices were constructed using VanRaden's first method, while transcriptomic relationship matrices were derived from miRNA and mRNA abundance data [3].
In plant studies, barley researchers implemented a cost-efficient RNA-Seq workflow utilizing small-footprint plant cultivation, high-throughput RNA extraction, and library preparation miniaturization [5]. This approach maintained data quality while reducing expenses, making transcriptomic prediction more accessible for breeding programs. Data processing typically includes quality control, read mapping, expression quantification, and normalization, with special attention to batch effects and technical variability.
Bioinformatics Workflow Solutions
Reproducible Workflow Management Systems
Modern bioinformatics workflows must balance reproducibility, portability, and computing platform independence while handling complex multi-omics data. Two predominant workflow systems have emerged as standards:
The Common Workflow Language (CWL) is a workflow description standard designed with a focus on portability, easy tool and workflow definitions, and reproducibility of data-intensive analysis workflows [60]. CWL relies on technologies including JSON-LD, Avro for data modeling, and Docker-compatible software container runtimes for portability. It has been adopted by leading institutions including the Wellcome Trust Sanger Institute and Institut Pasteur.
Nextflow is a workflow language and system that implements a domain-specific language built on Groovy [60]. It supports execution of workflows with partial resumption, containerization with Docker and Singularity, and multiple execution modes including local execution, execution on clusters, Amazon EC2, Kubernetes, and OpenStack. Nextflow adapts its execution strategy to the environment, providing exceptional flexibility across computing infrastructures.
Containerization for Enhanced Reproducibility
Containerization technologies such as Docker and Singularity are essential components of modern bioinformatics workflows, ensuring consistent software environments across heterogeneous computing platforms [60]. The H3ABioNet experience demonstrates that containerized workflows can successfully operate across diverse African computing environments, including High Performance Computing centers, university and lab clusters, and cloud environments [60]. This approach is particularly valuable for multi-omics prediction, where software dependencies and version compatibility present significant challenges to result reproducibility.
Diagram 1: Multi-omics prediction workflow architecture integrating data processing, workflow management, containerization, and statistical modeling
Research Reagent Solutions and Essential Materials
Key Computational Tools and Platforms
Table 3: Essential research reagents and computational tools for multi-omics prediction
| Tool/Platform | Category | Primary Function | Application in Multi-Omics Prediction |
|---|---|---|---|
| ASReml R [3] | Statistical Software | Fitting mixed linear models | Implementation of GBLUP, TBLUP, and related models for variance component estimation |
| CWL (Common Workflow Language) [60] | Workflow System | Portable workflow description | Reproducible execution of multi-omics data processing pipelines |
| Nextflow [60] | Workflow System | Scalable workflow execution | Distributed processing of large genomic and transcriptomic datasets |
| Docker [60] | Containerization | Environment reproducibility | Creating consistent software environments across computing platforms |
| Trimmomatic [60] | Data Preprocessing | Read trimming | Quality control of RNA-Seq data prior to expression quantification |
| BWA-MEM [60] | Read Alignment | Short read mapping | Alignment of RNA-Seq reads to reference genomes for variant calling |
| GATK [60] | Variant Calling | Variant discovery | Identifying SNPs from RNA-Seq data for consensus SNP datasets |
| Fluidigm BioMark HD [3] | Laboratory Platform | High-throughput genotyping | Candidate gene expression assessment for transcriptomic prediction |
The integration of transcriptomic data with genomic prediction models consistently demonstrates improved accuracy for complex traits, but requires careful attention to technical implementation details. The evidence from multiple studies indicates that model-based integration approaches such as GTCBLUPi outperform simple data concatenation, successfully addressing the redundancy between genomic and transcriptomic information layers [3] [17]. Successful implementation depends on robust bioinformatics workflows managed by systems like CWL or Nextflow, containerized using Docker or Singularity for reproducibility across heterogeneous computing environments [60]. For research groups embarking on multi-omics prediction, we recommend prioritizing workflow reproducibility and statistical models that explicitly account for inter-omics correlations to maximize prediction accuracy while maintaining computational tractability and biological interpretability.
Performance Comparison of Genomic Prediction Optimization Techniques
The integration of advanced optimization techniques significantly enhances the accuracy of genomic and transcriptomic prediction models. The table below summarizes quantitative performance gains from key studies.
Table 1: Performance Improvements from Optimization Techniques in Genomic Prediction
| Optimization Technique | Specific Method/Approach | Reported Performance Gain | Key Finding |
|---|---|---|---|
| Functional Annotation | SBayesRC (integrating 96 annotations) | 14% improvement in European ancestry; up to 34% in cross-ancestry prediction vs. non-annotation baseline [61]. | Integrates GWAS summary statistics with functional annotations to refine causal variant probability and effect distribution [61]. |
| Multi-Omics Integration | Model-based fusion (G+T) | Transcriptomic data explained a larger portion of phenotypic variance than host genetics for efficiency traits [3]. | Combines genomic (G) and transcriptomic (T) data, effectively complementing genetic information [3] [1]. |
| Feature Selection | SVM & Gradient Boosting with 1,000 pre-selected SNPs | Achieved a predictive accuracy (Spearman correlation) of 0.28 and 0.27, respectively, for residual feed intake in pigs [62]. | Using 500 or more SNPs selected via stable filter methods (e.g., spearcor, mrmr) yielded high accuracy and stability [62]. |
| Machine Learning Models | Non-parametric models (XGBoost, LightGBM) | +0.025 mean increase in Pearson's correlation (r) vs. Bayesian alternatives in multi-species benchmark [63]. | Offers modest accuracy gains and major computational advantages (faster fitting, lower RAM) [63]. |
Detailed Experimental Protocols
Protocol 1: Integrating Functional Annotations with SBayesRC
This protocol details the methodology for incorporating functional genomic annotations to improve polygenic prediction, as validated in large-scale human genetic studies [61].
- Input Data Requirements: The method requires two primary data sources:
- GWAS Summary Statistics: Effect sizes and standard errors from a genome-wide association study.
- Functional Annotations: A matrix of genomic annotations (e.g., from BaselineLD v2.2) across all SNPs. The original study used 96 functional annotations [61].
- LD Reference Panel: A reference sample, such as from the 1000 Genomes Project, to estimate linkage disequilibrium (LD) correlations between SNPs.
- Core Algorithmic Steps:
- Data Integration and LD Block Processing: GWAS summary statistics are integrated with the LD reference and functional annotations. The genome is divided into quasi-independent LD blocks, and a low-rank model based on eigen-decomposition is applied within each block to enhance computational efficiency and robustness to LD miscalibration [61].
- Annotation-Dependent Prior Application: The method uses a multicomponent, annotation-dependent mixture prior. This prior allows the functional annotations to influence both the probability of a SNP being causal and the distribution of its effect size, refining the signal from the GWAS data [61].
- Joint Parameter Estimation: All parameters, including SNP effects and annotation weights, are estimated jointly from the data using a full Bayesian learning framework, outputting posterior inclusion probabilities (PIP) and refined SNP effect estimates for polygenic score calculation [61].
- Validation Approach: Performance is typically assessed via cross-validation within a cohort or cross-biobank prediction, comparing the prediction R² of SBayesRC against baseline methods like SBayesR or LDpred2 that do not use functional annotations [61].
Protocol 2: Multi-Omics Integration with GTCBLUP
This protocol outlines the procedure for combining genomic and transcriptomic data while accounting for the redundancy between these layers, as implemented in a study on Japanese quail [3].
- Biological Material and Phenotyping: The experiment utilized an F₂ cross of 480 Japanese quails. Birds were raised under controlled conditions, and efficiency-related phenotypes (e.g., phosphorus utilization, body weight gain) were recorded during a strong growth phase [3].
- Omics Data Collection:
- Genotyping: Animals were genotyped using a 6k Illumina iSelect chip, resulting in 4k high-quality SNPs after standard quality control (e.g., minor allele frequency, call rate) [3].
- Transcriptomic Profiling: Ileum mucosa samples were collected at slaughter. RNA sequencing identified differentially expressed transcripts, and candidate miRNAs and mRNAs were quantified using a Fluidigm BioMark HD system [3].
- Statistical Modeling and Analysis:
- Data Transformation: Phenotypes were transformed using a Box-Cox transformation to approximate normal distributions [3].
- Model Comparison: Several models were fitted and compared:
- GBLUP: Uses only genomic relationship matrix (
G). - TBLUP: Uses only transcriptomic relationship matrix (
T). - GTBLUP: Uses both
GandTas independent random effects. - GTCBLUPi: The proposed model that integrates genomics with transcriptomics conditioned on genetics, effectively removing shared variation to avoid collinearity [3].
- GBLUP: Uses only genomic relationship matrix (
- Variance Component and Accuracy Estimation: Models were fitted using ASReml software to estimate the proportion of phenotypic variance explained by genomics and transcriptomics. Prediction accuracy was evaluated as the correlation between predicted and observed phenotypes in a cross-validation framework [3].
Protocol 3: Stable Feature Selection for Machine Learning
This protocol describes a framework for identifying a stable subset of predictive features (SNPs) for use with machine learning models, as applied to residual feed intake in pigs [62].
- Population and Phenotyping: The study used 5,708 boars from a terminal sire line. Individual daily feed intake was recorded using electronic feeders, and residual feed intake (RFI) was calculated as the residual from a regression of daily feed intake on average daily gain, backfat thickness, and metabolic weight [62].
- Genotyping and Quality Control: Animals were genotyped, and SNPs underwent standard quality control for minor allele frequency and call rate.
- Feature Selection and Modeling Pipeline:
- Application of FS Methods: A variety of FS methods were applied to the high-dimensional SNP data:
- Filter Methods: Univariate (
univ.dtree,spearcor) and multivariate (cforest,mrmr). - Embedded Methods: Elastic Net and LASSO regression.
- Combined Filters and Embedded Methods.
- Filter Methods: Univariate (
- Model Training with Selected Features: Different subsets of SNPs (e.g., 50 to 1,500) selected by the FS methods were used as input for learners, including Support Vector Machine (SVM) and Gradient Boosting (GB). The benchmark was GBLUP without pre-selection [62].
- Stability and Accuracy Assessment: A 10-fold cross-validation was performed. The stability of the FS methods was quantified using a specialized estimator that measures the agreement between feature subsets selected across different training folds, considering properties like correction for chance [62]. Accuracy was measured as the median Spearman correlation between observed and predicted RFI values across the cross-validation folds [62].
- Application of FS Methods: A variety of FS methods were applied to the high-dimensional SNP data:
Workflow and Relationship Visualizations
Multi-Omics Data Integration Workflow
The following diagram illustrates the logical flow and decision points in a robust multi-omics integration experiment, from data collection to model validation [3] [1].
Multi-Omics Integration Workflow
Feature Selection and Model Validation Logic
This diagram outlines the process of evaluating feature selection stability and its impact on the final prediction model's performance [62].
Feature Selection and Validation Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Genomic Prediction Optimization
| Item/Resource | Function in Research | Example from Literature |
|---|---|---|
| Fluidigm BioMark HD System | High-throughput microfluidic platform for targeted transcriptomic quantification (e.g., of candidate miRNAs/mRNAs) [3]. | Used for transcriptomic profiling of ileum mucosa in a quail model for efficiency traits [3]. |
| EasyGeSe Database | A curated collection of ready-to-use genomic and phenotypic datasets from multiple species for standardized benchmarking of prediction methods [63]. | Provides data from barley, maize, pig, rice, and others, enabling fair comparison of parametric vs. non-parametric models [63]. |
| GCTB Software | Software tool for implementing Bayesian models that integrate functional annotations, such as the SBayesRC method [61]. | Used for integrating 96 functional annotations with GWAS summary data to improve polygenic prediction [61]. |
| BaselineLD v2.2 Annotations | A curated set of functional genomic annotations from the Roadmap Epigenomics Project used to inform SNP priors [61]. | Served as the source of 96 functional annotations in the development and application of the SBayesRC method [61]. |
| PEREGGRN Benchmarking Platform | A software and dataset resource for standardized evaluation of gene expression forecasting methods against held-out genetic perturbations [64]. | Used to benchmark the accuracy of predicting transcriptomic changes from novel knockdown/overexpression experiments [64]. |
The field of genomics has experienced unprecedented data growth, driven by the widespread adoption of high-throughput sequencing technologies and the increasing complexity of multi-omics research. The exponential growth of genomics data poses a significant challenge for computing infrastructure and software algorithms for genomics analysis [65]. Genomic data analysis now routinely involves processing terabytes of data per project, with storage needs often requiring 100-200 times the input data size for temporary files during analysis [65]. This massive computational burden is particularly pronounced in transcriptomics and spatial genomics, where single-cell RNA sequencing and spatial transcriptomics platforms generate increasingly high-resolution data at subcellular levels [66] [67]. The convergence of increasing data volume, variety, and veracity creates a perfect storm that demands sophisticated computational strategies beyond traditional computing approaches. Researchers, scientists, and drug development professionals now face critical decisions in selecting appropriate computational infrastructures and platforms to handle these massive datasets efficiently while maintaining analytical rigor and accelerating discovery timelines for genomic and transcriptomic prediction models.
Cloud Computing Platforms for Genomic Data Analysis
Market Landscape and Key Providers
The cloud genomics market has expanded exponentially to address the computational challenges of modern genomic research. The market is anticipated to grow from $3.17 billion in 2024 to $3.91 billion in 2025, representing a compound annual growth rate (CAGR) of 23.3% [68]. This growth trajectory is expected to continue, with projections reaching $8.93 billion by 2029 [68]. Major cloud providers have established dedicated genomics services, with Amazon Web Services (AWS), Google Cloud Genomics, and Microsoft Azure emerging as dominant players offering specialized solutions for genomic data storage, processing, and analysis [14] [68]. These platforms provide scalable infrastructure that can handle vast datasets with ease, enabling global collaboration among researchers from different institutions who can work on the same datasets in real-time [14]. For smaller laboratories and research institutions, cloud computing offers cost-effective access to advanced computational tools without significant upfront infrastructure investments [14].
Technical Advantages for Genomic Workloads
Cloud computing platforms provide several technical advantages specifically designed for genomic workloads. They offer scalable storage and computational resources that can be dynamically allocated based on project needs, effectively handling the "bursty" nature of genomic analysis pipelines where intensive computation may be needed only during specific processing stages [65]. The scalability is particularly valuable for processing large-scale genomic datasets, such as those generated by the UK Biobank project, which already exceeds 50 TB for just 50,000 participants (approximately one-tenth of the total project) [65]. Additionally, cloud platforms comply with strict regulatory frameworks such as HIPAA and GDPR, ensuring secure handling of sensitive genomic data through advanced encryption, access controls, and audit trails [14]. These platforms also offer specialized genomic data services, such as Amazon Web Services' genomics-specific instances and Google Cloud's Life Sciences API, which provide optimized environments for running common genomic analysis tools and workflows [65].
Table 1: Comparison of Major Cloud Genomics Platforms
| Platform | Key Genomics Services | Unique Features | Use Case Strengths |
|---|---|---|---|
| Amazon Web Services (AWS) | AWS HealthLake, EC2 instances with high memory | X1e instances with up to 4 TB RAM and 128 cores | Large-scale population genomics, biobank analysis |
| Google Cloud | Google Cloud Genomics, Healthcare API | Integration with BigQuery for large-scale analytics | Collaborative research projects, machine learning on genomic data |
| Microsoft Azure | Azure Healthcare APIs, Genomics | Integration with Microsoft's research tools | Clinical genomics, integrated health solutions |
Deployment Models and Considerations
Cloud genomics solutions offer multiple deployment models to accommodate different research needs and regulatory requirements. Public clouds provide maximum scalability and cost-efficiency for non-sensitive research data, while private clouds offer dedicated resources for organizations with strict data governance requirements [68]. Hybrid cloud approaches are increasingly popular, allowing researchers to maintain sensitive data on-premises while leveraging cloud resources for computationally intensive analyses [68]. When selecting a cloud platform, researchers should consider data transfer costs for large genomic datasets, which can be substantial, and implement strategies such as data compression and selective transfer of processed rather than raw data. Performance optimization also requires careful selection of instance types, with memory-optimized instances (such as AWS's X1e instances with up to 4 TB of RAM) particularly valuable for genome assembly and other memory-intensive operations [65].
Scalable Analysis Frameworks and Architectures
High-Performance Computing (HPC) Approaches
Traditional high-performance computing clusters remain a powerful solution for scalable genomics analysis, particularly for institutions with existing HPC infrastructure. Message Passing Interface (MPI) has emerged as the de facto industry standard for distributed memory systems, enabling parallelism across multiple compute nodes [65]. MPI-based genomic tools, including read aligners like pBWA and assemblers like Ray, have demonstrated scalability up to hundreds of thousands of cores on HPC clusters [65]. The Partitioned Global Address Space (PGAS) programming model represents an alternative approach that combines advantages of shared-memory programming with the performance of message passing [65]. UPC++ implementations have shown remarkable performance in challenging genomic problems, with tools like Meta-HipMer successfully assembling a 2.6 TB metagenome dataset in just 3.5 hours using 512 nodes [65]. While HPC approaches deliver exceptional performance for suitable algorithms, they require significant expertise in parallel programming and face challenges in fault tolerance, where failure of one process can lead to failure of the entire application [65].
Big Data Frameworks for Genomics
Big data frameworks originally developed for web-scale applications have been adapted to address genomic analysis challenges. The Hadoop framework, with its Hadoop Distributed File System (HDFS) and MapReduce programming model, enables load-balanced, scalable, and robust solutions for big data analytics [65]. Several Hadoop-based applications have been developed for genomics, including specialized tools for NGS read alignment, genetic variant calling, and sequence analysis [65]. However, the Input/Output-intensive nature of Hadoop's MapReduce can severely limit performance for genomic workflows, as map tasks often produce 10-100× the amount of intermediate data stored in local disks until reduce tasks fetch them, creating significant communication overhead [65]. Apache Spark has gained popularity as an alternative that improves upon Hadoop's limitations by keeping intermediate results in memory, making it particularly suitable for iterative machine learning algorithms commonly used in genomic prediction models [69].
Specialized Hardware Accelerators
Specialized processing units have emerged to improve the efficiency of parallel genomic computation. Field-Programmable Gate Arrays (FPGAs) can provide remarkable acceleration for specific genomic algorithms, with Falcon Computing developing an FPGA-based solution that speeds up the Genome Analysis Tool Kit (GATK) by 50 times [65]. Graphics Processing Units (GPUs) have a long history in computational biology and are increasingly applied to NGS data analysis, particularly for deep learning applications in genomics [65]. Tensor Processing Units (TPUs) represent another specialized architecture gaining traction for genomic deep learning applications [65]. While these specialized hardware architectures can dramatically accelerate specific computations, they present limitations including availability, difficulty scaling on heterogeneous systems, and the need to port existing CPU-based algorithms to these specialized systems [65]. Additionally, training large deep neural networks on GPUs/TPUs can be cost-prohibitive for some research organizations [65].
Performance Comparison of Scalable Platforms
Benchmarking Scalable Frameworks for Transcriptomics
Several studies have systematically evaluated the performance of scalable frameworks for transcriptomics analysis. The Dask framework has emerged as a particularly efficient solution for parallelizing Python-based genomic analysis workflows. Dask divides data into smaller blocks on which to perform highly parallel computations, allowing larger datasets to fit in the memory of single machines while tightly integrating with existing Python data analytics libraries [69]. This approach minimizes the need for code rewrites and facilitates transition to HPC environments, making it especially valuable for transcriptomics predictive modeling [69]. Benchmark studies comparing Dask to traditional Hadoop and Spark frameworks have demonstrated its advantages for medium to large-scale transcriptomics analyses, particularly for iterative machine learning tasks such as hyperparameter optimization and nested cross-validation [69]. These tasks become computationally prohibitive with traditional frameworks due to their input/output overhead but can be efficiently parallelized with Dask's task-scheduling approach [69].
Table 2: Performance Comparison of Scalable Analysis Frameworks
| Framework | Parallelization Strategy | Maximum Demonstrated Scale | Genomics Applications | Key Limitations |
|---|---|---|---|---|
| MPI-based Tools | Message passing across distributed memory systems | Hundreds of thousands of cores | pBWA, Ray, genome assembly | Complex programming model, poor fault tolerance |
| Hadoop/MapReduce | Data partitioning with disk-based shuffling | Thousands of nodes | Variant calling, sequence analysis | High I/O overhead, unsuitable for iterative algorithms |
| Apache Spark | In-memory data processing with resilient distributed datasets | Thousands of nodes | Variant calling, machine learning | Memory management complexity, steep learning curve |
| Dask | Dynamic task scheduling with blocked algorithms | Thousands of cores | Transcriptomics, machine learning, general Python workflows | Younger ecosystem, less mature for some genomic applications |
Case Study: Scalable Machine Learning for Transcriptomics
Machine learning pipelines for transcriptomics analysis present particular computational challenges that benefit from specialized scalable approaches. A standard supervised learning workflow for gene expression data includes data loading and preprocessing, train/test splitting, model training with cross-validation, hyperparameter optimization, and model evaluation [69]. The computational demands are amplified by the need for feature selection, scaling, and normalization of noisy RNA-seq expression data with large amplitude variation [69]. The combination of k-fold cross-validation and hyperparameter optimization into nested cross-validation, while providing exhaustive performance estimates, creates particularly intensive computational requirements [69]. In benchmark studies, Dask has demonstrated efficient parallelization of these workflows, seamlessly integrating with popular Python machine learning libraries like scikit-learn while handling the large dimensionality of transcriptomics data, where the number of features (genes) often far exceeds the number of samples [69]. The framework's ability to work with larger-than-memory datasets through blocked algorithms and intelligent scheduling makes it particularly suitable for single-cell transcriptomics studies, which routinely profile 10,000+ cells per sample [70].
Experimental Protocols for Benchmarking Computational Platforms
Standardized Benchmarking Methodology
Rigorous benchmarking of computational platforms requires standardized experimental protocols to ensure fair comparison across different systems. A comprehensive approach should include multiple dataset sizes (from small-scale pilot studies to population-scale datasets), diverse genomic workflows (including read alignment, variant calling, transcript quantification, and genome assembly), and systematic monitoring of computational metrics including execution time, memory usage, storage I/O, scalability, and cost efficiency [65] [69]. For cloud platforms, benchmarking should evaluate both vertical scaling (increasing resources on a single node) and horizontal scaling (adding more nodes to a cluster), as optimal configurations vary significantly across different genomic analysis tasks [65]. The benchmarking protocol should also assess ease of implementation, including setup time, code modification requirements, and learning curve, as these factors significantly impact real-world adoption and productivity [65] [69].
Diagram: Benchmarking Methodology for Genomic Computational Platforms. This workflow outlines the systematic approach for evaluating the performance of different computational platforms for genomic analysis.
Reference Datasets and Benchmarking Tools
Well-characterized reference datasets are essential for standardized benchmarking of genomic computational platforms. The Genome in a Bottle (GIAB) consortium provides benchmark variant call sets that serve as gold standards for evaluating variant detection pipelines [65]. For transcriptomics analysis, the SEQC/MAQC-III consortium has established comprehensive RNA-seq reference datasets with validated expression profiles [69]. The Spatial Transcriptomics benchmarking studies provide standardized datasets for comparing performance across different spatial genomics platforms, incorporating adjacent tissue sections analyzed by complementary technologies like CODEX and single-cell RNA sequencing to establish ground truth data [66] [67]. Specialized benchmarking tools such as GIAB's hap.py for variant calling evaluation and SpatialOMICs for spatial transcriptomics platform comparison provide standardized metrics and methodologies [66]. These reference resources enable reproducible benchmarking across different computational platforms and help researchers select optimal infrastructure for their specific analytical needs.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Computational Platforms and Tools for Scalable Genomic Analysis
| Category | Specific Tools/Platforms | Primary Function | Key Considerations |
|---|---|---|---|
| Cloud Platforms | AWS Genomics, Google Cloud Life Sciences, Azure Genomics | Scalable infrastructure for genomic workflows | Data transfer costs, compliance requirements, instance selection |
| HPC Frameworks | MPI, OpenMP, PGAS/UPC++ | Parallel computing on clusters | Programming complexity, fault tolerance, resource scheduling |
| Big Data Frameworks | Hadoop, Spark, Dask | Distributed data processing | Learning curve, memory management, integration with bioinformatics tools |
| Specialized Hardware | GPUs (NVIDIA), FPGAs, TPUs | Accelerated computing for specific algorithms | Cost, specialized programming requirements, algorithm compatibility |
| Workflow Management | Nextflow, Snakemake, Cromwell | Pipeline orchestration and reproducibility | Portability, monitoring capabilities, resource optimization |
| Benchmarking Tools | GIAB benchmarks, SpatialOMICs, hap.py | Performance evaluation and validation | Reference data quality, metric selection, reproducibility |
The computational demands of modern genomic and transcriptomic research require careful strategic selection of appropriate computing platforms based on specific research objectives, dataset characteristics, and organizational constraints. Cloud computing platforms offer unparalleled scalability and accessibility, particularly for projects with variable computational needs or limited local infrastructure [14] [68]. High-performance computing clusters provide maximum performance for suitable algorithms and established workflows, especially for institutions with existing HPC investments [65]. Specialized frameworks like Dask offer compelling advantages for Python-centric transcriptomics analysis and machine learning workflows, seamlessly integrating with popular analytical libraries while enabling scaling from single machines to large clusters [69]. As genomic technologies continue to evolve toward even higher resolution, including subcellular spatial transcriptomics and whole-genome sequencing at population scale, the strategic importance of computational platform selection will only increase. Researchers and drug development professionals should prioritize platforms that not only address current analytical needs but also provide flexible pathways for scaling as data volumes and analytical complexity continue their exponential growth trajectory.
Improving Model Interpretability and Generalization Across Diverse Datasets
This guide objectively compares the performance of models using only genomic data against those incorporating transcriptomic data for phenotypic prediction, a key focus in modern multi-omics research.
Integrating transcriptomic data with traditional genomic information consistently enhances prediction accuracy for complex traits across multiple species. The performance gain is particularly notable for traits closely linked to metabolic processes and in cross-population prediction scenarios.
Table 1: Summary of Key Multi-Omic Prediction Performance Findings
| Study & Organism | Trait Category | Best Performing Model | Key Performance Finding |
|---|---|---|---|
| Japanese Quail [3] [16] | Efficiency (e.g., Phosphorus Utilization) | GTCBLUPi (Integrated Genomic & Transcriptomic) | Transcriptome explained a larger portion of phenotypic variance than host genetics alone. |
| Barley RIL Populations [5] | Complex Agronomic Traits | Consensus SNP (RNA-Seq + WGS) | Surpassed 50K SNP array benchmark; advantage most prominent in inter-population predictions. |
| Drosophila melanogaster [71] | Starvation Resistance | Variable Selection Methods (using expression) | Achieved higher accuracy than models using only genotype data. |
| Alfalfa [38] | Salt Tolerance | Multi-omics GS (GWAS + RNA-seq) | Improved prediction accuracy, enabling more precise selection for a complex polygenic trait. |
Detailed Experimental Protocols & Data
Japanese Quail Efficiency Traits Study
This experiment provides a formal framework for integrating transcriptomic data while accounting for redundancy with genomic information [3] [16].
- Experimental Population: 480 F2 cross Japanese quails (Coturnix japonica) from a larger founder population, raised under controlled conditions [3].
- Phenotyping: Measured efficiency-related traits including phosphorus utilization (PU), body weight gain (BWG), feed intake (FI), and feed conversion ratio (FCR) [3].
- Omics Data Collection:
- Statistical Analysis:
Barley Multi-Parent RIL Population Study
This study demonstrated the cost-effective use of RNA-Seq data for genomic prediction in a plant breeding context [5].
- Genetic Material: 237 Recombinant Inbred Lines (RILs) from three connected spring barley sub-populations (HvDRR13, HvDRR27, HvDRR28) [5].
- Plant Cultivation & RNA Extraction:
- Library Preparation & Sequencing: Miniaturized VAHTS Universal V6 RNA-seq Library Prep, sequenced on Illumina platforms [5].
- Data Processing & Analysis:
- RNA-Seq Data Processing Pipeline:
Performance Data and Model Interpretability
Quantitative Prediction Accuracies
Table 2: Detailed Prediction Accuracies Across Models and Datasets
| Organism | Trait | Genomic-Only Model Accuracy | Transcriptomic-Only Model Accuracy | Integrated Model Accuracy | Notes |
|---|---|---|---|---|---|
| Barley [5] | 8 Agronomic Traits | ~0.73-0.78 (50K SNP array) | Similar to or better than SNP array (RNA-Seq data) | ~0.73-0.78 (Consensus SNP) | Consensus SNP (RNA-Seq + WGS) performed best, with significant gains in inter-population prediction. |
| Drosophila [71] | Starvation Resistance (Females) | Lower than transcriptomic | Varies by method | Highest with Variable Selection Methods | Method performance dependent on trait architecture; variable selection worked well for starvation resistance. |
| Drosophila [71] | Startle Response | Lower than transcriptomic | Varies by method | Lower with Variable Selection | Highlights the need for method selection based on trait. |
| Japanese Quail [3] | Efficiency Traits | Lower than integrated models | Higher than genomic data | Highest with GTCBLUPi | Transcript abundances explained larger phenotypic variance portion than genetics. |
Insights into Model Interpretability
- Biological Mechanism Insight: In the Japanese quail study, models using transcriptomic data allowed for the estimation of "transcriptomic correlations" between traits and the identification of polygenic backgrounds based on transcriptomic profiles, offering deeper biological insight than genomic markers alone [3] [16].
- Gene Function Validation: The alfalfa salt tolerance study demonstrated that integrating GWAS with RNA-seq data enabled robust candidate gene identification and functional validation, moving beyond simple prediction to mechanistic understanding [38].
- Trait-Relevant Feature Selection: Research on Drosophila showed that incorporating functional annotation (Gene Ontology) into prediction models could improve accuracy and highlight biologically significant genes and pathways, such as the Insulin-like Receptor (InR) for starvation resistance [71].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for Multi-Omic Prediction
| Reagent / Platform | Function / Application | Example Use Case |
|---|---|---|
| Illumina iSelect Chip [3] | High-throughput genomic SNP genotyping. | Generating 4k SNP dataset in Japanese quail study [3]. |
| Fluidigm BioMark HD System [3] [16] | High-throughput microfluidic PCR for targeted gene expression analysis. | Profiling candidate miRNA and mRNA transcripts in quail subpopulations [3] [16]. |
| VAHTS Universal V6 RNA-seq Library Prep Kit [5] | Preparation of Illumina-compatible mRNA sequencing libraries. | Cost-effective, miniaturized library construction in barley RNA-Seq study [5]. |
| TRIzol Reagent [5] | Monophasic solution for RNA isolation from cells and tissues. | High-throughput, 96-well format total RNA extraction from barley seedlings [5]. |
| ASReml-R Software [3] | Statistical software for fitting linear mixed models using REML. | Fitting GBLUP, TBLUP, and GTCBLUP models for variance component estimation and prediction [3]. |
Discussion and Research Implications
The consistent outperformance of integrated models, particularly for complex traits, underscores that transcriptomic data captures a portion of phenotypic variance that is complementary to, and sometimes greater than, that captured by genomics alone [3] [5] [71]. The GTCBLUPi model provides a critical framework for this integration by explicitly conditioning transcriptomic data on genomics to avoid collinearity and isolate non-genetic effects [3] [16].
For cross-population prediction and breeding applications, the use of RNA-Seq data is particularly promising. It not only provides gene expression information but can also be used to call genetic variants, creating a powerful consensus dataset when combined with parental WGS data [5].
Future research should focus on standardizing cost-effective, high-throughput omics data collection protocols and developing more sophisticated, yet user-friendly, modeling frameworks that can capture non-linear and hierarchical interactions between different omics layers to fully realize the potential of multi-omics prediction [17].
Rigorous Benchmarking and Performance Metrics Across Domains
This guide provides an objective comparison of the performance between genomic and transcriptomic prediction models, synthesizing quantitative evidence from recent scientific studies. It is structured to help researchers and drug development professionals evaluate these models based on key metrics: prediction accuracy, Root Mean Square Error (RMSE), and R-squared values.
Quantitative Performance Comparison of Prediction Models
The table below summarizes the quantitative performance of various prediction models across different species and traits, as reported in recent research.
Table 1: Performance Metrics of Genomic and Transcriptomic Prediction Models
| Study & Organism | Traits | Model | Key Performance Metric | Reported Value | Performance Notes |
|---|---|---|---|---|---|
| Japanese Quail [3] [16] | Phosphorus Utilization, Body Weight Gain, Feed Efficiency | GBLUP (Genomic) | Portion of Phenotypic Variance Explained | Lower than transcriptomic models | Genomics alone explained a smaller portion of variance [3]. |
| TBLUP (Transcriptomic) | Portion of Phenotypic Variance Explained | Larger than genomic models | Transcript abundances explained a larger portion of phenotypic variance [3]. | ||
| GTBLUP (Combined) | Prediction Accuracy | Highest | Combining both data types resulted in the highest prediction accuracies [3] [16]. | ||
| Barley [55] | Agriculturally Important Traits | RNA-Seq SNP Data (Transcriptomic) | Prediction Ability (5-fold CV) | 0.73 - 0.78 | Demonstrated that transcriptomic data alone can effectively predict complex traits [55]. |
| Consensus SNP (Genomic + Transcriptomic) | Prediction Ability | 0.73 - 0.78 (5/8 traits significantly better than 50K SNP array) | Performance was most prominent in inter-population predictions [55]. | ||
| Dairy Cattle (Simulated) [72] | Complex Traits | GBLUP (Genomic) | Predictive Ability (Baseline) | Baseline | Used as a benchmark for comparison [72]. |
| WMKRR (Genomic + Predicted Transcriptomic) | Predictive Ability | +1.12% to +3.23% over GBLUP | Improvement varied based on feature selection scenario [72]. | ||
| Dairy Cattle (Real) [72] | Complex Traits | GBLUP (Genomic) | Predictive Ability (Baseline) | Baseline | Used as a benchmark for comparison [72]. |
| WMKRR (Genomic + Predicted Transcriptomic) | Predictive Ability | +4.66% to +8.41% over GBLUP | Superior performance in both cross- and forward-validation [72]. | ||
| Multiple Crops [73] | Various Agronomic Traits | LSTM (Deep Learning on Genomic Data) | STScore (Performance Metric) | 0.967 (Average across 6 datasets) | Superior performance in capturing additive and epistatic effects [73]. |
Detailed Experimental Protocols
To ensure reproducibility and provide context for the data in Table 1, this section details the methodologies from key cited experiments.
Protocol: Multi-Omics Integration in Japanese Quail
This experiment systematically compared the variance explained and prediction accuracy of models using genomic data, transcriptomic data, and their combination [3] [16].
- Objective: To estimate the proportion of phenotypic variance explained by transcripts and genomic markers, and to evaluate the predictive performance of several BLUP models [3].
- Population: 480 F2 cross Japanese quails (Coturnix japonica) raised under controlled conditions [3] [16].
- Phenotypes: Efficiency-related traits including phosphorus utilization (PU), body weight gain (BWG), feed intake (FI), and feed conversion ratio (FCR). Phenotypes were Box-Cox transformed for analysis [3] [16].
- Genotyping: Performed using a 6k Illumina iSelect chip, resulting in 4k SNPs after filtering [3] [16].
- Transcriptomics: Ileum mucosa samples were collected. miRNA and mRNA candidates were assessed using 96.96 dynamic arrays on a Fluidigm BioMark HD system [3] [16].
- Models Tested:
- GBLUP:
y = Xb + Zg*g + e(Genomic data only) [3]. - TBLUP:
y = Xb + Zt*t + e(Transcriptomic data only) [3]. - GTBLUP:
y = Xb + Zg*g + Zt*t + e(Both genomic and transcriptomic data as independent effects) [3]. - GTCBLUPi:
y = Xb + Zg*g + Zc*tc + e(Transcriptomic data conditioned on genetic effects to remove redundant information) [3].
- GBLUP:
- Analysis: Models were executed using ASReml R software. Variance components and prediction accuracies were compared [3].
Protocol: Transcriptomic Prediction in Barley RILs
This study evaluated the capability of low-cost RNA-Seq data to perform genomic prediction in barley recombinant inbred lines (RILs), comparing it to traditional SNP arrays [55].
- Objective: To investigate the potential of RNA-Seq data for genomic prediction and to increase prediction ability by combining genomic and transcriptomic datasets [55].
- Genetic Material: 237 spring barley RILs from three related sub-populations (HvDRR13, HvDRR27, HvDRR28) [55].
- Phenotypes: Eight agriculturally important traits measured in up to seven environments [55].
- RNA Sequencing:
- Plant Cultivation: Seedlings were grown in controlled, miniaturized conditions for seven days [55].
- RNA Extraction: Total RNA was extracted from 50 mg of plant material using a miniaturized TRIzol protocol in a 96-well format [55].
- Library Prep: mRNA libraries were constructed using a miniaturized VAHTS Universal V6 RNA-seq Library Prep Kit for Illumina [55].
- Data Types for Prediction:
- Gene expression dataset from RNA-Seq.
- SNP dataset called from RNA-Seq data.
- Consensus SNP dataset between RNA-Seq and parental Whole-Genome Sequencing (WGS) data.
- A 50K SNP array was used as a benchmark [55].
- Validation: A fivefold cross-validation scheme was used to evaluate prediction ability. Inter-population predictions were also performed [55].
Model Workflow and Performance Visualization
The following diagram illustrates the typical workflow for developing and evaluating a multi-omics prediction model, integrating the key steps from the experimental protocols above.
Figure 1: Workflow for developing and evaluating multi-omics prediction models. The process begins with data collection and preprocessing, followed by parallel training of different model types. Quantitative evaluation consistently shows that combined models (red arrow) achieve superior predictive performance.
The quantitative findings from multiple studies are synthesized in the chart below, providing a clear visual comparison of model performance.
Figure 2: Comparative performance of genomic, transcriptomic, and combined prediction models. The synthesis of recent studies reveals a consistent pattern: models integrating both genomic and transcriptomic data achieve the highest predictive performance, followed by transcriptomic-only models, with genomic-only models serving as a baseline.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 2: Key Reagents and Platforms for Genomic and Transcriptomic Studies
| Item | Function / Application | Example Use Case |
|---|---|---|
| Illumina iSelect Chip | High-throughput SNP genotyping to obtain genomic markers. | Genotyping of 480 Japanese quails, yielding 4k filtered SNPs [3] [16]. |
| Fluidigm BioMark HD System | High-throughput microfluidic platform for targeted gene expression analysis. | Profiling of 77 miRNA and 80 mRNA candidates in quail ileum samples [3] [16]. |
| VAHTS Universal V6 RNA-seq Library Prep Kit | Library preparation for RNA sequencing on Illumina platforms. | Construction of mRNA libraries from barley RILs in a miniaturized protocol [55]. |
| TRIzol Reagent | Monophasic solution for the isolation of high-quality total RNA from cells and tissues. | RNA extraction from 50 mg of barley plant material in a 96-well format [55]. |
| ASReml R Software | Statistical software for fitting linear mixed models using REML, commonly used for GBLUP and variants. | Execution of GBLUP, TBLUP, and GTBLUP models in the Japanese quail study [3]. |
| XGBoost Algorithm | A machine learning algorithm based on gradient-boosted decision trees, effective for tabular data. | Used for gene selection and reconstruction of full transcriptomic signals from a reduced gene set [74]. |
The accurate prediction of complex traits and disease outcomes is a cornerstone of modern biological research and precision medicine. For years, genomic data has been the primary source for building predictive models, leveraging an individual's DNA sequence to forecast phenotypes. More recently, transcriptomic data, which captures dynamic gene expression patterns, has emerged as a complementary predictive source. The integration of these and other biological data layers, known as multi-omics, promises a more comprehensive view of the biological system. However, a critical question remains: does this integration consistently yield a measurable improvement in predictive performance over single-omics approaches?
This guide provides a direct, evidence-based comparison of the predictive performance of genomics-only, transcriptomics-only, and multi-omics models. By synthesizing recent experimental findings across diverse fields—from plant and animal breeding to clinical disease prediction—we aim to offer researchers a clear understanding of the relative strengths and limitations of each approach. The accompanying data, methodologies, and resource toolkit are designed to inform the strategic design of future predictive studies.
Performance Data at a Glance
The following tables consolidate key quantitative findings from recent studies, offering a direct comparison of model performance across different omics approaches and biological contexts.
Table 1: Performance Comparison in Crop and Livestock Studies
| Study Organism & Trait | Genomics-Only Model (Accuracy) | Transcriptomics-Only Model (Accuracy) | Multi-Omics Model (Accuracy) | Key Metric |
|---|---|---|---|---|
| Chinese Simmental Cattle (Meat Quality Traits) [75] | GBLUP: Baseline | TBLUP: Lower than GBLUP | MBLUP: +3.37% to 4.18% over GBLUP | Predictive Accuracy |
| Rice (Hybrid Yield) [75] | Baseline Predictability | Not Reported | Metabolomics Integration: ~2x Increase vs. Genomics | Predictability |
| Alfalfa (Salt Tolerance) [38] | Standard GS Models | Not a standalone model | GWAS + RNA-seq + ML: Enhanced precision | Precision & Selection Efficiency |
Table 2: Performance Comparison in Clinical Prediction Studies
| Study & Prediction Target | Genomics-Only Model (Performance) | Transcriptomics-Only Model (Performance) | Multi-Omics Model (Performance) | Key Metric |
|---|---|---|---|---|
| Preterm Birth (PTB) Prediction [76] | cfDNA Model: AUC 0.822 | cfRNA Model: AUC 0.851 | Integrated cfDNA+cfRNA: AUC 0.89 | AUC (Area Under Curve) |
| Breast Cancer Subtyping [39] | Not a standalone model | Not a standalone model | MOFA+ (Multi-omics): F1 Score 0.75 | F1 Score (Classification) |
Detailed Experimental Protocols and Methodologies
To ensure the reproducibility of these comparative analyses, this section outlines the core experimental and computational methodologies employed in the cited studies.
Kernel-Based Multi-Omics Integration in Beef Cattle
A 2022 study on Chinese Simmental beef cattle provided a clear framework for integrating genomic and transcriptomic data using kernel-based methods [75].
- Data Collection: Phenotypes for meat quality traits (e.g., shear force, water holding capacity) were recorded. Genotyping was performed using the Illumina BovineHD BeadChip (770,000 SNPs), and transcriptomic data was obtained via RNA sequencing of longissimus dorsi muscle tissue.
- Model Construction: The researchers used the Cosine kernel to transform both SNP and gene expression data into
n x nsymmetric relationship matrices (G-matrix and T-matrix). They defined five prediction models:- GBLUP: Utilized only the genomic relationship matrix.
- TBLUP: Utilized only the transcriptomic relationship matrix.
- MBLUP: Combined matrices linearly:
M = ratio * G + (1-ratio) * T. - mssBLUP & wmssBLUP: Extended the model to integrate a larger genotyped population with a smaller transcribed subset.
- Evaluation: Predictive accuracy was calculated as the correlation between the genomic estimated breeding values (GEBVs) and the observed phenotypes. The multi-omics models (MBLUP, wmssBLUP) consistently outperformed the single-omics GBLUP model across all traits, demonstrating the value of transcriptomic data [75].
Transformer-Based Multi-Omics Model for Preterm Birth
A 2025 study on preterm birth (PTB) prediction showcased the application of a deep learning architecture for multi-omics integration in a clinical context [76].
- Data Collection: The study involved 682 pregnant women. Cell-free DNA (cfDNA) and cell-free RNA (cfRNA) were isolated from plasma samples and sequenced.
- Model Construction: The team developed GeneLLM, a gene-focused large language model based on a Transformer architecture, to interpret the complex patterns in the sequencing data. They built and compared three models:
- A cfDNA-only model (representing genomics).
- A cfRNA-only model (representing transcriptomics).
- An integrated model combining cfDNA and cfRNA data.
- Evaluation: Model performance was evaluated using the Area Under the Receiver Operating Characteristic Curve (AUC). The integrated model achieved a significantly higher AUC (0.89) compared to either single-omics model, with the transcriptomics-only model also outperforming the genomics-only approach [76].
Statistical vs. Deep Learning Multi-Omics Integration in Breast Cancer
A 2025 study on breast cancer subtyping directly compared two common approaches for multi-omics integration: a statistical method and a deep learning method [39].
- Data: The analysis integrated three omics layers from 960 patient samples: host transcriptomics, epigenomics (methylation), and microbiomics.
- Integration Methods:
- MOFA+ (Statistical): An unsupervised tool that uses factor analysis to infer a set of latent factors that capture the common variance across all omics datasets.
- MoGCN (Deep Learning): A graph convolutional network that uses autoencoders to reduce dimensionality and learn features for subtype classification.
- Evaluation Protocol: After integration, the top 100 features from each omics layer were selected from each model. These feature sets were then used to train and evaluate separate classifiers (Support Vector Classifier and Logistic Regression) to predict breast cancer subtypes. Performance was assessed using the F1 score to account for class imbalance.
- Findings: The statistical-based MOFA+ model outperformed the deep learning-based MoGCN, achieving a higher F1 score (0.75) and identifying a greater number of biologically relevant pathways [39].
Visualizing Multi-Omics Workflows and Performance
The following diagrams illustrate the core workflows for multi-omics integration and summarize the performance relationships identified in the research.
Multi-Omics Integration Workflow
Performance Relationship Diagram
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful multi-omics research relies on a suite of wet-lab and computational tools. The table below lists key resources referenced in the studies.
Table 3: Key Reagents and Computational Tools for Multi-Omics Research
| Item Name | Function/Application | Relevance to Multi-Omics Studies |
|---|---|---|
| Illumina BovineHD BeadChip [75] | High-density genotyping of single nucleotide polymorphisms (SNPs). | Provides high-quality genomic marker data for constructing genomic relationship matrices in genomic prediction models. |
| Cell-free DNA (cfDNA) & RNA (cfRNA) [76] | Non-invasive sampling of genetic material from blood plasma. | Enables clinical prediction models (e.g., for preterm birth) without invasive tissue biopsies. |
| Cosine Kernel Transformation [75] | A mathematical function to map high-dimensional omics data into a similarity matrix. | Converts genomic and transcriptomic features into n x n relationship matrices (G and T) compatible with BLUP models. |
| MOFA+ (Multi-Omics Factor Analysis) [39] | A statistical tool for unsupervised integration of multi-omics data. | Identifies latent factors that drive variation across different omics datasets, aiding in dimensionality reduction and feature selection. |
| Graph Convolutional Network (GCN) [39] | A type of deep neural network that operates on graph-structured data. | Used in methods like MoGCN to integrate multi-omics data and learn patterns for tasks like cancer subtyping. |
| Transformer Architecture [76] | A deep learning model architecture, foundational for large language models. | Adapted in models like GeneLLM to interpret sequential biological data from multiple omics layers for enhanced prediction. |
The collective evidence from recent studies demonstrates a clear and consistent trend: multi-omics models frequently achieve superior predictive performance compared to models based on a single omics layer. The performance gain of multi-omics integration is observed across diverse fields, from agriculture to clinical medicine, and is robust across different modeling techniques, including kernel-based methods, statistical factor analysis, and deep learning.
While transcriptomics-only models can, in some cases, outperform genomics-only models—as seen in preterm birth prediction—their performance can be variable and trait-dependent. Genomics remains a powerful and stable predictor, particularly for highly heritable traits. However, by complementing the static genetic blueprint with dynamic information from the transcriptome, multi-omics models capture a more complete picture of the biological processes leading to a phenotype, leading to more accurate and biologically informed predictions. Researchers should consider a multi-omics strategy, with careful attention to the choice of integration method, to maximize the predictive power of their studies.
In the field of modern breeding, accurately predicting complex traits is a cornerstone for accelerating genetic gain. While genomic selection, which uses DNA markers to predict breeding values, has been a revolutionary tool, its accuracy is often limited for traits governed by intricate biological pathways. In recent years, transcriptomic data, which captures gene expression patterns, has emerged as a powerful complementary source of information. This case study provides a objective performance comparison between traditional genomic prediction models and emerging transcriptomic and multi-omics approaches. We synthesize experimental data from recent studies across plant and animal species to offer breeders and researchers a clear guide on the relative merits, applicable protocols, and essential tools for implementing these strategies.
Performance Comparison: Genomics vs. Transcriptomics vs. Multi-Omics
Recent research consistently demonstrates that integrating multiple layers of biological information enhances predictive accuracy. The tables below summarize key performance metrics from recent, authoritative studies.
Table 1: Predictive Performance in Animal Breeding (Japanese Quail Study)
| Prediction Model | Data Types Used | Key Performance Findings | Reference |
|---|---|---|---|
| GBLUP | Genomic (SNPs) | Served as a baseline for genomic prediction. | [3] [16] |
| TBLUP | Transcriptomic (mRNA/miRNA) | Explained a larger portion of phenotypic variance than genomics alone for efficiency-related traits. | [3] [16] |
| GTBLUP | Genomic + Transcriptomic | Outperformed models using only one data type, but with collinearity challenges. | [3] [16] |
| GTCBLUPi | Genomic + Conditioned Transcriptomic | Achieved the highest prediction accuracy by effectively handling redundancy between data layers. | [3] [16] |
Table 2: Predictive Performance in Plant Breeding (Multi-Species Studies)
| Species | Genomic-Only Model | Transcriptomic/Multi-Omics Model | Performance Change | Reference |
|---|---|---|---|---|
| Maize | GBLUP | Model-based Multi-Omics Integration | Consistent improvement, especially for complex traits. | [1] [17] |
| Barley | 50k SNP Array | RNA-Seq Consensus SNP Dataset | Prediction abilities of 0.73-0.78; significantly better for 5/8 traits. | [5] |
| Rice | GBLUP | Multi-Omics (G+T+M) with Deep Learning | Performance gains varied with trait complexity and modeling approach. | [1] [17] |
| General | Single GBLUP Model | Ensemble of Multiple Prediction Models | Increased accuracies and reduced prediction errors. | [77] |
Detailed Experimental Protocols
To ensure reproducibility and provide a clear understanding of the cited performance data, this section details the methodologies from the key experiments.
Animal Breeding Protocol: Japanese Quail Efficiency Traits
The following workflow outlines the experimental and analytical procedures from the Japanese quail study [3] [16].
1. Experimental Population and Design: The study used an F2 cross of 480 Japanese quails raised under controlled conditions. Birds were housed in metabolism units and fed a diet designed to let them express their full genetic potential for phosphorus utilization (PU) [3] [16].
2. Phenotyping: The measured efficiency-related traits included:
- Phosphorus Utilization (PU): Based on total P intake and excretion (%).
- Body Weight Gain (BWG): Measured between days 10 and 15 (g).
- Feed Intake (FI): Recorded over a 5-day period (g).
- Feed Conversion Ratio (FCR): Calculated as FI divided by BWG (g/g).
- Tibia Ash (TA): Total amount of tibia ash (mg).
- Calcium Utilization (CaU): Based on total Ca intake and excretion (%) [3] [16].
3. Genotyping and Transcriptomics: Animals were genotyped using a 6k Illumina iSelect chip, resulting in 4k SNPs after quality control. For transcriptomics, ileum mucosa samples were collected. The top differentially expressed transcripts were identified and quantified using 96.96 dynamic arrays on a Fluidigm BioMark HD system [3] [16].
4. Statistical Analysis:
- Data Preprocessing: Phenotypes were transformed using a Box-Cox transformation to achieve normality and then scaled and centered [3] [16].
- Models Tested:
- GBLUP: Used only genomic relationship matrix.
- TBLUP: Used only transcriptomic relationship matrix (separate models for miRNA and mRNA).
- GTBLUP: Combined genomic and transcriptomic effects as independent random effects.
- GTCBLUPi: Integrated genomic data with transcriptomic data conditioned on the genotypes to remove shared variation and avoid collinearity [3] [16].
- Evaluation: Models were compared based on the proportion of phenotypic variance explained ((\sigma^2g/\sigma^2p)) and the accuracy of predicting phenotypes [3] [16].
Plant Breeding Protocol: Multi-Omics in Maize and Rice
The following workflow outlines the procedures from the multi-omics plant studies [1] [5] [17].
1. Plant Materials and Phenotyping: Studies utilized diverse panels of inbred lines or Recombinant Inbred Lines (RILs). For example:
- Maize282: 279 lines with 22 traits.
- Maize368: 368 lines with 20 traits.
- Rice210: 210 lines with 4 traits. Phenotyping was conducted under single-environment conditions to isolate the effect of omics integration without genotype-by-environment interactions [1] [17].
2. Multi-Omics Data Generation:
- Genomics (G): High-density SNP markers (e.g., 50,878 to 100,000 markers).
- Transcriptomics (T): RNA sequencing (RNA-Seq) data providing thousands of gene expression features.
- Metabolomics (M): Metabolic profiles quantifying hundreds to thousands of metabolites [1] [17].
3. Data Integration and Modeling: A wide array of integration strategies was evaluated.
- Early Fusion: Simple concatenation of omics datasets.
- Model-Based Fusion: Advanced methods (e.g., Bayesian, Deep Learning) capable of capturing non-additive and hierarchical interactions between omics layers.
- Ensemble Methods: Combining predictions from multiple individual models to reduce error [1] [77] [17].
4. Validation: Predictive performance was rigorously assessed using fivefold cross-validation. Some studies also tested inter-population prediction, where the training and validation sets came from different genetic populations, to evaluate model robustness [5].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successfully implementing the protocols above requires a suite of specialized reagents and platforms. The following table lists key solutions used in the featured studies.
Table 3: Key Research Reagent Solutions for Genomic and Transcriptomic Prediction
| Research Solution | Specific Example | Function in Workflow | Reference |
|---|---|---|---|
| Genotyping Array | Illumina iSelect Chip (6k) | High-throughput genotyping to obtain genome-wide SNP markers. | [3] [16] |
| RNA Extraction Kit | TRIzol Reagent | Isolation of high-quality total RNA from tissue samples (e.g., ileum, seedling). | [5] |
| RNA-Seq Library Prep Kit | VAHTS Universal V6 RNA-seq Library Prep Kit | Preparation of sequencing-ready libraries from mRNA for transcriptome profiling. | [5] |
| High-Throughput qPCR System | Fluidigm BioMark HD System | Targeted quantification of candidate mRNA and miRNA transcript abundances. | [3] [16] |
| NGS Platform | Illumina NovaSeq X, Oxford Nanopore | Whole-genome sequencing and RNA-Seq; provides long reads and high throughput. | [14] |
| Statistical Software | ASReml R, R Studio | Fitted mixed linear models (e.g., GBLUP) for variance component and accuracy estimation. | [3] [16] |
The empirical data presented in this guide leads to several key conclusions for professionals in the field. First, transcriptomic data often explains a larger proportion of phenotypic variance than genomic data alone, as it captures the dynamic state of gene expression closer to the functional level of the trait [3] [16]. Second, while combining data types is powerful, the modeling approach is critical. Naive concatenation can underperform, whereas sophisticated models like GTCBLUPi or model-based multi-omics fusion that account for data redundancy and interaction effects deliver the most consistent accuracy gains [3] [1]. Finally, the "best" model is context-dependent. For traits with strong additive genetic effects, GBLUP remains robust. For complex traits with non-additive effects, deep learning or ensemble methods that leverage diverse predictions show significant promise [77] [78].
In conclusion, the integration of transcriptomic data with genomics represents a significant step forward in the accurate prediction of complex traits. Breeders should consider a tiered strategy: beginning with established genomic selection and progressively incorporating transcriptomic layers and advanced modeling for high-value, complex traits where greater predictive accuracy justifies the additional cost and complexity.
The accurate prediction of drug response and binding affinity is a cornerstone of modern drug discovery and personalized medicine. It enables the identification of effective therapeutic candidates and the anticipation of patient-specific treatment outcomes, thereby streamlining the development pipeline and improving clinical success rates. Current computational approaches largely leverage two key types of biological information: genomic data, which provides a static blueprint of an organism's genetic makeup, and transcriptomic data, which captures the dynamic expression of genes, offering a snapshot of cellular activity. This case study objectively compares the performance of prediction models built on these two data types, synthesizing recent evidence to guide researchers and drug development professionals in selecting the most appropriate methodologies for their work. The analysis reveals that while both data types are valuable, transcriptomic information often provides a more direct and powerful correlate of phenotypic outcome.
Performance Comparison: Genomic vs. Transcriptomic Models
Quantitative comparisons across multiple independent studies consistently demonstrate that models incorporating transcriptomic data frequently achieve superior prediction accuracy for drug response and related complex traits compared to those relying solely on genomic information.
Table 1: Comparison of Prediction Accuracy for Complex Traits
| Trait / Context | Genomic Model Performance | Transcriptomic Model Performance | Integrated Model Performance | Citation |
|---|---|---|---|---|
| Efficiency Traits (Japanese Quail) | GBLUP: Explained lower phenotypic variance | TBLUP: Explained larger portion of phenotypic variance | GTBLUPi: Highest prediction accuracy | [3] |
| Agricultural Traits (Barley) | 50K SNP Array: Served as benchmark | RNA-Seq SNP Data: Achieved comparable or better prediction ability | Consensus SNP Dataset: Best performance, especially in inter-population predictions | [5] |
| Drug Response (Cancer Cell Lines) | Models using mutation & CNV: Lower performance | Gene Expression Models: Superior performance (SVR best algorithm) | Integration of mutation & CNV did not improve predictions | [79] |
| Personal Gene Expression | Enformer, Basenji2, etc.: Limited performance explaining variation across individuals | N/A | Models often failed to predict correct direction of effect of genetic variants | [80] |
The superiority of transcriptomic data is attributed to its closer proximity to the phenotypic outcome. As noted in the quail study, transcript abundances "provide insights into gene expression patterns, which are shaped by both genetic and environmental factors, offering a more comprehensive understanding of the expression of phenotypes" [3]. Similarly, in plant breeding, the transcriptome is recognized as "a promising predictor, bridging the gap between the genome and the trait" [5].
Experimental Protocols and Methodologies
Protocol for Multi-Omics Prediction in Animal Breeding
A study on Japanese quail provides a robust framework for comparing genomic and transcriptomic predictions, using Best Linear Unbiased Prediction (BLUP) models [3].
- Biological Material: 480 F2 cross Japanese quails (Coturnix japonica) raised under controlled conditions.
- Phenotyping: Measured efficiency-related traits including phosphorus utilization (PU), body weight gain (BWG), feed intake (FI), and feed conversion ratio (FCR).
- Genotyping: Conducted using a 6k Illumina iSelect chip, resulting in 4k filtered SNPs. The genomic relationship matrix (G) was calculated following VanRaden's first method [3].
- Transcriptomic Profiling: Ileum mucosa samples were collected. miRNA and mRNA sequencing identified differentially expressed transcripts. Abundances of 77 miRNAs and 80 mRNAs were quantified for 482 quails using Fluidigm BioMark HD system [3].
- Statistical Models:
- GBLUP:
y = Xb + Zg*g + e(Uses only genomic random effects) - TBLUP:
y = Xb + Zt*t + e(Uses only transcriptomic random effects) - GTBLUPi:
y = Xb + Zg*g + Zct*c + e(Integrates both, conditioning transcripts to address redundancy) [3]
- GBLUP:
- Data Analysis: All analyses were executed using ASReml R in R Studio. Phenotypes were transformed using a Box-Cox transformation to address skewed distributions [3].
Protocol for Drug Response Prediction in Cancer Cell Lines
Research utilizing the GDSC dataset offers a standard protocol for predicting drug response using machine learning [79].
- Data Source: Genomic profiles and IC50 drug response values for 734 cancer cell lines from the GDSC database.
- Feature Sets:
- Gene Expression: Matrix of 734 cell lines × 8,046 genes.
- Mutation Data: Binary matrix (734 × 636) indicating presence/absence of mutations.
- Copy Number Variation (CNV): Binary matrix (734 × 694) indicating CNV status.
- Feature Selection: Methods tested included Mutual Information (MI), Variance Threshold (VAR), Select K Best (SKB), and a biologically informed list from the LINCS L1000 dataset (627 genes) [79].
- Regression Algorithms: 13 algorithms from scikit-learn were compared, including Support Vector Regression (SVR), Elastic Net, Random Forests, and Multi-layer Perceptron (MLP) [79].
- Model Training & Evaluation: Performance was evaluated using a 3-fold cross-validation, with Mean Absolute Error (MAE) as the primary metric [79].
Workflow for Multi-Omics Prediction
The following diagram illustrates the logical workflow for developing and comparing genomic and transcriptomic prediction models, as applied in the case studies above.
Key Challenges and Limitations
Despite the promising performance of transcriptomic models, several significant challenges remain that impact the accuracy and generalizability of both genomic and transcriptomic predictions.
Data Bias and Leakage in Binding Affinity Prediction
A critical issue in protein-ligand binding affinity prediction is the inflation of performance metrics due to data leakage between training and test sets. A 2025 study revealed that nearly half of the complexes in the common CASF benchmark shared exceptionally high structural similarity with complexes in the PDBbind training database, allowing models to "perform comparably well... after omitting all protein or ligand information" [81]. This indicates that impressive benchmark results were often driven by memorization and exploitation of structural similarities rather than a genuine understanding of protein-ligand interactions. The proposed solution, "PDBbind CleanSplit," rigorously filters the training data to eliminate these similarities, providing a more realistic assessment of model generalizability [81].
Failure in Predicting Direction of Effect
State-of-the-art genomic deep learning models (e.g., Enformer, Basenji2), while successful at predicting gene expression levels from the reference genome, show limited performance in explaining expression variation across individuals based on personal genomic sequences [80]. Alarmingly, when these models do detect regulatory variation, they often fail to predict the correct direction of effect of cis-regulatory genetic variants on expression. This shortcoming persists even for genes with strong genetic associations, highlighting a fundamental gap in current models' ability to interpret personal genome variation [80].
Benchmarking of Expression Forecasting
A comprehensive benchmarking of methods that forecast gene expression changes in response to genetic perturbations found that it is uncommon for these methods to outperform simple baselines [64]. This benchmarking platform, PEREGGRN, evaluated methods on 11 large-scale perturbation datasets and highlighted the importance of using a nonstandard data split where no perturbation condition occurs in both training and test sets to avoid illusory success [64].
The Scientist's Toolkit
The following table details essential reagents, datasets, and software solutions used in the featured experiments, providing a resource for researchers seeking to implement these methodologies.
Table 2: Key Research Reagent Solutions and Materials
| Item Name | Type | Function/Application | Example Use Case |
|---|---|---|---|
| GDSC Database | Dataset | Provides genomic profiles & drug sensitivity (IC50) data for cancer cell lines for training ML models. | Drug response prediction [82] [83] [79] |
| PDBbind Database | Dataset | A comprehensive collection of protein-ligand complexes with binding affinity data for training scoring functions. | Binding affinity prediction [84] [81] |
| Fluidigm BioMark HD | Instrument | High-throughput microfluidic system for quantifying transcript abundances (e.g., mRNA, miRNA). | Transcriptomic profiling in quail study [3] |
| LINCS L1000 Dataset | Dataset/Gene Set | Provides a list of ~1,000 landmark genes that show significant response in drug screens; used for feature selection. | Filtering informative genes for drug response prediction [79] |
| PDBbind CleanSplit | Dataset | A curated version of PDBbind with reduced data leakage, enabling genuine evaluation of model generalization. | Robust binding affinity model training [81] |
| Scikit-learn | Software Library | Provides accessible implementations of 13+ regression algorithms (SVR, Random Forest, etc.). | Drug response prediction [79] |
| ASReml R | Software | Statistical software used for fitting mixed linear models, including GBLUP and related multi-omics models. | Genomic and transcriptomic prediction in animal breeding [3] |
| TRIzol Reagent | Chemical | A ready-to-use reagent for the isolation of high-quality total RNA from cells and tissues. | RNA extraction from barley seedlings [5] |
This comparative analysis leads to several key conclusions for researchers and drug development professionals. The evidence strongly indicates that transcriptomic data often serves as a more powerful predictor for drug response and complex traits than genomic data alone, as it captures dynamic biological states closer to the phenotype. Furthermore, the most robust prediction strategies frequently involve the integration of multiple data types using models specifically designed to handle their redundancy, such as GTCBLUPi.
However, the field must contend with significant challenges, including pervasive data bias in public benchmarks and the limited ability of current deep learning models to accurately predict the effects of personal genetic variation. Moving forward, the adoption of rigorously filtered datasets, like PDBbind CleanSplit, and the development of more biologically grounded models are imperative to improve the generalizability and real-world impact of predictive models in drug discovery and personalized medicine.
Analysis of Influential Covariates and Features in High-Performing Models
The accurate prediction of complex traits is a fundamental objective in genetics, crucial for accelerating genetic gain in plant and animal breeding and for advancing personalized therapeutic strategies in medicine. For years, genomic prediction (GP) models, which utilize genome-wide molecular markers, have been the cornerstone of this effort [85]. However, the sole reliance on genomic data often fails to fully capture the intricate biological pathways that lead to phenotypic expression. Consequently, attention has shifted towards transcriptomic data, which provides a dynamic snapshot of gene expression patterns, offering a closer link to the eventual phenotype [3] [86].
This guide provides an objective comparison of genomic and transcriptomic prediction models, framing the analysis within the broader thesis that multi-omics integration is key to unlocking higher predictive performance. We will dissect the influential covariates and experimental factors that differentiate top-performing models, supported by quantitative data and detailed methodologies from recent studies. The analysis is intended for researchers, scientists, and drug development professionals who require a clear, evidence-based overview of the current state and future directions of predictive modeling in genetics.
Performance Comparison: Genomic vs. Transcriptomic Models
Direct comparisons between genomic and transcriptomic prediction models reveal a nuanced landscape where the best-performing approach often depends on the trait's genetic architecture, the available data, and the biological context.
Quantitative Trait Prediction in Plants
Studies in plant breeding consistently demonstrate that transcriptome-based prediction is a powerful alternative or complement to traditional genomics.
Table 1: Comparison of Prediction Accuracies in Rice Using Different Data Types
| Trait | Trait Category | Prediction Model | Accuracy (R²) | Key Finding |
|---|---|---|---|---|
| Crown Root Diameter [87] | Root Phenotype | Transcriptomic (All Genes) | 0.59 | Root-specific transcripts significantly improved prediction. |
| Transcriptomic (Root-Specific Genes) | 0.66 | |||
| Crown Root Length [87] | Root Phenotype | Genomic Prediction | Not Reported | Root transcripts were more accurate than leaf transcripts for root traits. |
| Transcriptomic (Leaf) | Lower Accuracy | |||
| Transcriptomic (Root) | Higher Accuracy | |||
| Plant Height [87] | Shoot Phenotype | Transcriptomic (Leaf) | Higher Accuracy | Leaf transcripts were more accurate than root transcripts for shoot traits. |
| Transcriptomic (Root) | Lower Accuracy |
A study on rice accessions found that using tissue-specific transcripts markedly improved prediction for traits associated with that tissue. For instance, models using root transcripts predicted root phenotypes like crown root length more accurately than models using leaf transcripts, and vice versa for shoot phenotypes like plant height [87]. Furthermore, selecting gene subsets based on biological knowledge, such as using only root-specifically expressed genes, boosted the predictive accuracy for crown root diameter by over 10% compared to using all genes [87].
Integrated Models for Complex Traits
While transcriptomic data alone can be highly predictive, the most robust models often integrate multiple omics layers to account for shared and unique sources of variation.
Table 2: Model Performance for Efficiency-Related Traits in Japanese Quail
| Model Type | Model Description | Key Finding on Phenotypic Variance | Overall Performance |
|---|---|---|---|
| GBLUP [3] | Uses genomic data only. | Explained a smaller portion of variance. | Baseline performance. |
| TBLUP [3] | Uses transcriptomic data only. | Transcripts from ileum tissue explained a larger portion of variance than genetics. | Outperformed GBLUP. |
| GTBLUP [3] | Simple combination of genomic and transcriptomic data. | -- | Suffered from collinearity between data layers. |
| GTCBLUPi [3] | Integrated model correcting for redundancy. | Effectively partitioned genetic and non-genetic effects. | Highest prediction accuracy. |
Research on efficiency-related traits in Japanese quail demonstrated that transcript abundances from intestinal tissue explained a larger portion of phenotypic variance than host genetics alone [3]. Models that incorporated both genetic and transcriptomic information, particularly the GTCBLUPi model which was specifically designed to address redundant information between the omics layers, outperformed models using only one data type [3]. This confirms that transcriptomic information effectively complements genetic data, but requires sophisticated statistical integration to avoid collinearity.
Experimental Protocols and Methodologies
The reliability of model comparisons hinges on rigorous and reproducible experimental designs. Below are detailed protocols from key studies cited in this guide.
Protocol 1: Multi-Omics Integration for Genomic Prediction
This protocol is based on the study "Genomic prediction powered by multi-omics data" [17].
- Objective: To evaluate the performance of 24 different integration strategies combining genomic (G), transcriptomic (T), and metabolomic (M) data for predicting complex traits in plants.
- Datasets: Three public datasets (Maize282, Maize368, Rice210) containing genotypic, transcriptomic, metabolomic, and phenotypic data for continuous agronomic traits.
- Data Preprocessing:
- Genotyping: SNPs were filtered for quality and minor allele frequency.
- Transcriptomics: RNA-seq data was normalized (e.g., FPKM, TPM).
- Metabolomics: Metabolite abundances were log-transformed and scaled.
- Integration Strategies:
- Early Fusion: Simple concatenation of features from different omics layers into a single input matrix.
- Model-Based Fusion: Use of statistical and machine learning models (e.g., Bayesian models, deep learning) capable of capturing non-additive and hierarchical interactions between omics layers.
- Model Training & Evaluation:
- Models were trained using a cross-validation scheme to ensure that the performance was evaluated on unseen genotypes.
- Predictive accuracy was measured using the Pearson correlation between observed and predicted phenotypic values.
- Key Findings: Model-based fusion methods consistently outperformed genomic-only models and simple concatenation approaches, particularly for complex traits.
Protocol 2: Transcriptome-Based Prediction with Gene Subsets
This protocol is based on the study "Transcriptome-based prediction for polygenic traits in rice using different gene subsets" [87].
- Objective: To test whether selecting gene subsets based on a priori biological knowledge can improve the predictive ability of transcriptome-based models for polygenic traits.
- Biological Material: 57 diverse rice accessions from the World Rice Core Collection (WRC).
- Phenotyping: Plants were grown in an upland field, and both shoot (e.g., plant height, tiller number) and root (e.g., crown root length, root dry weight) traits were measured.
- Transcriptome Profiling:
- RNA was extracted from both leaf and root tissues and pooled across three biological replicates per accession.
- RNA-seq libraries were sequenced, and read counts were normalized to FPKM values. Expression level was defined as log₂(FPKM + 1).
- Gene Subset Selection: Genes were partitioned into subsets based on three features:
- Tissue Specificity: Genes specifically expressed in leaves or roots.
- Ontology Annotations: Genes associated with specific Gene Ontology (GO) terms.
- Co-expression Modules: Genes belonging to the same co-expression network module.
- Modeling and Validation:
- Prediction models were trained using the expression levels of genes within a selected subset.
- Predictive ability (R²) was calculated using cross-validation and compared against the baseline model that used all expressed genes.
Signaling Pathways and Workflows
The following diagrams illustrate the logical workflow for comparing prediction models and the conceptual process of multi-omics integration, which underpin the experimental protocols discussed.
Model Comparison Workflow
Diagram Title: Model Comparison Workflow
Multi-Omics Data Integration
Diagram Title: Multi-Omics Data Integration
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the experimental protocols requires a suite of reliable reagents, platforms, and analytical tools.
Table 3: Key Research Reagent Solutions for Genomic and Transcriptomic Studies
| Item Name | Function/Application | Specific Examples/Notes |
|---|---|---|
| Next-Generation Sequencer | High-throughput sequencing of genomes (DNA) and transcriptomes (RNA). | Illumina NovaSeq X series (for large-scale projects); Oxford Nanopore platforms (for long-read, real-time sequencing) [14]. |
| RNA-seq Library Prep Kit | Preparation of RNA samples for sequencing, converting RNA into a library of cDNA fragments with adapters. | Kits are often platform-specific (e.g., Illumina TruSeq, Nanopore cDNA-PCR Sequencing Kit). Critical for generating high-quality transcriptomic data [87]. |
| Genotyping Array | Genome-wide profiling of single nucleotide polymorphisms (SNPs). | Illumina iSelect chip (e.g., used in the quail study with a 6k array [3]). Cost-effective for large breeding populations. |
| Fluidigm BioMark HD System | High-throughput microfluidic platform for targeted gene expression analysis. | Used for validating RNA-seq findings and profiling pre-selected candidate genes (e.g., miRNAs and mRNAs) in a large number of samples [3]. |
| Statistical Software & Packages | Fitting statistical models for genomic and transcriptomic prediction. | ASReml-R (for mixed linear models like GBLUP [3]), R/Bioconductor packages (e.g., edgeR for RNA-seq normalization [87]), and custom Python/R scripts for machine learning. |
| Cloud Computing Platform | Providing scalable computational resources for storing and analyzing large omics datasets. | Amazon Web Services (AWS), Google Cloud Genomics, and Microsoft Azure are essential for handling multi-terabyte projects and enabling collaboration [14]. |
Cross-Validation and Inter-Population Prediction Robustness
In the evolving field of genomic selection, the integration of transcriptomic data with traditional genomic information has emerged as a promising approach to enhance phenotypic prediction accuracy. However, the true test of any prediction model lies not just in its performance within a single population, but in its ability to generalize across diverse genetic backgrounds—a capability known as inter-population prediction robustness. This robustness is precisely quantified through rigorous cross-validation frameworks that simulate how models perform when applied to genetically distinct groups.
Cross-validation provides an essential methodology for evaluating model performance by partitioning data into training and validation sets, allowing researchers to estimate how well their models will generalize to unseen data. When applied to inter-population predictions, these validation techniques become crucial for assessing whether models trained on one genetic population can accurately predict traits in another. For researchers, scientists, and drug development professionals, understanding these validation paradigms is fundamental to deploying reliable predictive models in both agricultural and biomedical contexts.
This guide systematically compares the performance of genomic versus transcriptomic prediction models across multiple studies, with particular emphasis on their inter-population prediction capabilities as validated through robust cross-validation frameworks. We present quantitative comparisons, detailed methodological protocols, and practical research tools to inform model selection for various prediction tasks.
Performance Comparison: Genomic vs. Transcriptomic Prediction Models
Quantitative Analysis of Prediction Performance Across Species
Table 1: Comparative performance of genomic and transcriptomic prediction models
| Study Organism | Model Type | Prediction Accuracy | Inter-Population Performance | Key Findings |
|---|---|---|---|---|
| Japanese Quail [3] [16] | GBLUP (Genomic) | Baseline | Not reported | Explained smaller portion of phenotypic variance |
| TBLUP (Transcriptomic) | Higher than GBLUP | Not reported | Transcript abundances explained larger portion of phenotypic variance | |
| GTBLUP (Combined) | Highest | Not reported | Combination of both data types resulted in highest prediction accuracy | |
| Barley RIL Populations [55] [5] | RNA-Seq Data Only | 0.73-0.78 | Moderate | Capable of predicting complex traits alone |
| 50K SNP Array | Lower than RNA-Seq | Poorer | Benchmark; performed worse in inter-population predictions | |
| Consensus SNP Dataset | 0.73-0.78 | Highest | Best inter-population performance; advantage most prominent across RIL sub-populations |
Inter-Population Prediction Robustness Analysis
The most compelling evidence for transcriptomic data's value comes from inter-population validation studies. In barley research, the critical test occurred when models trained on one recombinant inbred line (RIL) sub-population were used to predict traits in different RIL sub-populations [55] [5]. This stringent validation approach revealed that the consensus SNP dataset derived from RNA-Seq and parental whole-genome sequencing data significantly outperformed traditional 50K SNP arrays.
The advantage of transcriptomic-informed models was "most prominent in the inter-population predictions, in which the training and validation sets originated from different RIL sub-populations" [55] [5]. This finding demonstrates that transcriptomic data can capture biological information that transcends population-specific genetic architectures, potentially reflecting conserved functional pathways rather than population-specific marker patterns.
Experimental Protocols and Methodologies
Cross-Validation Frameworks for Robustness Assessment
Table 2: Cross-validation methods for assessing prediction robustness
| Validation Method | Implementation | Advantages | Limitations | Suitable Scenarios |
|---|---|---|---|---|
| K-Fold Cross-Validation [88] [89] | Dataset split into K equal folds; model trained on K-1 folds and validated on remaining fold | Reduces overfitting; uses complete dataset | Choice of K affects performance estimate; computationally expensive than hold-out | General model performance assessment |
| Five-Fold Cross-Validation [55] [5] | Specific case with K=5; 80% training, 20% validation in each iteration | Balance between computational cost and reliability | May still have variance in performance estimates | Used in barley transcriptomic prediction study |
| Stratified K-Fold [89] | Maintains class distribution proportions across folds | Essential for imbalanced datasets | More complex implementation | Classification problems with class imbalance |
| Repeated K-Fold [89] | Multiple runs of K-fold with different random splits | More robust performance estimate | Computationally intensive | Final model evaluation when sufficient resources |
| Hold-Out Validation [89] | Single split into training and test sets | Simple and fast implementation | High variance; dependent on single split | Large datasets; initial model prototyping |
Statistical Models for Genomic-Transcriptomic Integration
The studies employed sophisticated statistical models to integrate genomic and transcriptomic data while accounting for inherent collinearity:
GBLUP (Genomic Best Linear Unbiased Prediction): This baseline model uses genomic relationship matrices derived from SNP data to predict breeding values [3] [16]. The genomic relationship matrix (G) was computed following VanRaden's first method [3] [16].
TBLUP (Transcriptomic BLUP): Similar to GBLUP but uses transcript abundance data instead of SNPs to construct relationship matrices, capturing similarities based on gene expression patterns [16].
GTBLUP: Integrates both genomic and transcriptomic data as independent random effects in a combined model, though this approach may face collinearity issues due to overlapping information between data layers [3].
GTCBLUP/GTCBLUPi: Advanced models that address redundancy between genomic and transcriptomic information by conditioning transcriptomic effects on genetic effects, ensuring the transcriptomic components capture purely non-genetic variation [3] [16]. This approach follows the Perez et al. method [3] [16] to handle the challenge that "using both SNP genotypes and other omics data as independent random effects in a mixed linear model leads to collinearity problems."
Visualization of Experimental Workflows and Conceptual Frameworks
Cross-Validation Workflow for Inter-Population Prediction
Cross-Validation Framework for Inter-Population Prediction: This diagram illustrates the process of rigorously validating prediction models across different genetic populations, where models trained on some populations are tested on excluded populations to assess generalizability.
Integrated Genomic-Transcriptomic Prediction Framework
Integrated Multi-Omics Prediction Framework: This workflow illustrates the pipeline from raw genomic and transcriptomic data through integrated modeling and rigorous validation, highlighting the critical role of cross-validation in assessing model robustness.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential research reagents and platforms for genomic-transcriptomic prediction studies
| Reagent/Platform | Function | Example Implementation |
|---|---|---|
| Illumina iSelect Chip [3] [16] | Genotyping platform for SNP discovery | Used for genotyping 480 Japanese quails with 6k SNPs filtered to 4k SNPs |
| Fluidigm BioMark HD System [3] [16] | High-throughput gene expression analysis | Assessed miRNA and mRNA candidates in quail study using 96.96 dynamic arrays |
| TRIzol Reagent [55] [5] | RNA extraction and purification | Used for high-throughput RNA extraction from barley seedlings in 96-well format |
| VAHTS Universal V6 RNA-seq Library Prep Kit [55] [5] | Library preparation for RNA sequencing | Constructed full-length mRNA libraries with miniaturized reagent volumes (25% of original) |
| Poly-A Tail mRNA Capture [55] [5] | mRNA selection from total RNA | Isolated mRNA using poly-A tail capture method with 1μg total RNA input |
| ASReml R Software [3] [16] | Statistical analysis of mixed linear models | Implemented BLUP models for variance component estimation and prediction |
| NanoPhotometer NP 80 [55] [5] | Nucleic acid quantification | Measured total RNA concentration after extraction |
The comparative analysis of genomic and transcriptomic prediction models reveals several critical insights for researchers and drug development professionals. First, transcriptomic data consistently explains a larger portion of phenotypic variance than genomic data alone across multiple species [3] [16]. Second, integrated models that combine both genomic and transcriptomic information generally outperform models using either data type independently. Third, and most significantly, transcriptomic-informed models demonstrate superior robustness in inter-population prediction scenarios, which is the most rigorous test of model generalizability [55] [5].
For practitioners designing prediction studies, the recommendation is clear: incorporate transcriptomic data whenever possible, especially when predictions need to generalize across diverse populations. The GTCBLUPi framework provides a robust statistical approach for integrating these data types while accounting for collinearity [3] [16]. Furthermore, five-fold cross-validation with intentional inter-population validation splits emerges as a critical methodology for producing reliable estimates of real-world performance.
As multi-omics technologies continue to become more accessible, the integration of transcriptomic data with traditional genomic approaches represents a promising path toward more accurate and robust predictive models in both agricultural and biomedical contexts.
Conclusion
The comparative analysis reveals that transcriptomic data often accounts for a larger portion of phenotypic variance than genomic data alone, providing a more dynamic and functional view of biological systems. However, the most accurate and robust prediction models strategically integrate both genomic and transcriptomic information, leveraging their complementary strengths. Methodologies that address data redundancy, such as the GTCBLUPi framework, and those employing advanced machine learning, are proving most effective. Future progress hinges on overcoming challenges related to data standardization, computational complexity, and model interpretability. The integration of emerging technologies like AI, single-cell sequencing, and spatial transcriptomics will further refine these models, accelerating advancements in precision medicine, drug discovery, and sustainable agricultural breeding. The choice between genomic, transcriptomic, or integrated models should be guided by the specific trait complexity, available resources, and desired biological insights.
References
- 1. Genomic prediction powered by multi-omics data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12485622/]
- 2. Genomic Selection in the Era of Next Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5186759/]
- 3. Incorporating transcriptomic data into genomic prediction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12551188/]
- 4. Incorporating gene expression and environment for ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1506434/full]
- 5. Assessment of genomic prediction capabilities ... [https://link.springer.com/article/10.1007/s00122-025-05029-0]
- 6. Comparison of genomic prediction accuracies in dairy cattle ... [https://gsejournal.biomedcentral.com/articles/10.1186/s12711-025-00966-2]
- 7. EasyGeSe – a resource for benchmarking genomic prediction ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12129-0]
- 8. Multi-omics: a bridge connecting genotype and phenotype for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12175374/]
- 9. Novel Functional Genomics Approaches Bridging ... [https://www.sciencedirect.com/science/article/pii/S2667174322000921]
- 10. Chromosome-Scale Genome and Comparative ... [https://pubmed.ncbi.nlm.nih.gov/34712262/]
- 11. Genomic and transcriptomic variation defines the ... [https://www.nature.com/articles/s42003-020-01377-3]
- 12. Valuable lessons-learned in transcriptomics experimentation [https://pmc.ncbi.nlm.nih.gov/articles/PMC4581358/]
- 13. Genomic & Transcriptomic Data Visualization [https://www.mll.com/en/genomic-transcriptomic-data-visualization]
- 14. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 15. What is the difference between genomics and ... [https://synapse.patsnap.com/article/what-is-the-difference-between-genomics-and-transcriptomics]
- 16. Incorporating transcriptomic data into genomic prediction ... [https://gsejournal.biomedcentral.com/articles/10.1186/s12711-025-01008-7]
- 17. Genomic prediction powered by multi-omics data [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1636438/full]
- 18. eQTLs are key players in the integration of genomic and... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9238188/]
- 19. Transcriptome analysis reveals novel insights into the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511390/]
- 20. Transcriptomic analysis unveils critical genes governing ... [https://www.sciencedirect.com/science/article/abs/pii/S1744117X25001790]
- 21. Transcriptomics technologies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5436640/]
- 22. Experimental and Data Analysis Planning for RNA-Seq [https://www.lexogen.com/blog/rna-lexicon-experimental-and-data-analysis-planning-for-rna-sequencing/]
- 23. Transcriptome analysis [https://www.illumina.com/techniques/multiomics/transcriptomics.html]
- 24. Comparison of the Efficiency of BLUP and GBLUP in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7142406/]
- 25. Comparison on genomic predictions using three GBLUP ... [https://gsejournal.biomedcentral.com/articles/10.1186/1297-9686-44-8]
- 26. Comparison of genomic predictions using ... [https://www.sciencedirect.com/science/article/pii/S0022030214005591]
- 27. Transcriptome prediction performance across machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8087249/]
- 28. Incorporating transcriptomic data into genomic prediction ... [https://pubmed.ncbi.nlm.nih.gov/41131481/]
- 29. Deep learning-driven drug response prediction and ... [https://www.nature.com/articles/s41598-025-91571-2]
- 30. AI Atlas #15: Ensemble Methods in Deep Learning [https://glasswing.vc/blog/ai-atlas-15-ensemble-methods-in-deep-learning/]
- 31. What is ensemble learning? [https://www.ibm.com/think/topics/ensemble-learning]
- 32. Deep neural network prediction of genome-wide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8866467/]
- 33. Comparing 3 Ensemble Learning Algorithms (Machine ... [https://medium.com/@smriithhiii/comparing-3-ensemble-learning-algorithms-machine-learning-9acd46e3be09]
- 34. Deep-learning-based gene perturbation effect prediction ... [https://www.nature.com/articles/s41592-025-02772-6]
- 35. Genomic Predicted cross performance: a tool for optimizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12620651/]
- 36. Genomics and multiomics in the age of precision medicine [https://pmc.ncbi.nlm.nih.gov/articles/PMC12106083/]
- 37. Multimodal fusion strategies for survival prediction in breast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12595345/]
- 38. Integrative multi-omics and genomic prediction reveal ... [https://www.sciencedirect.com/science/article/abs/pii/S1673852725002358]
- 39. Comparative analysis of statistical and deep learning-based ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06662-5]
- 40. CropARNet: A deep learning framework for crop genomic ... [https://www.sciencedirect.com/science/article/pii/S2772899425000242]
- 41. Deep learning methods for drug response prediction in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC9975164/]
- 42. Comparative evaluation of feature reduction methods for ... [https://www.nature.com/articles/s41598-024-81866-1]
- 43. A drug response prediction method for single-cell tumors ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1631898/full]
- 44. A drug response prediction method for single-cell tumors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12408664/]
- 45. Single-Cell Sequencing: Genomic and Transcriptomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11901077/]
- 46. Spatial Transcriptomics Market Size 2025 to 2034 [https://www.precedenceresearch.com/spatial-transcriptomics-market]
- 47. Systematic comparison of sequencing-based spatial ... [https://www.nature.com/articles/s41592-024-02325-3]
- 48. a novel precision medicine technique in nephrology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8137400/]
- 49. A practical guide for choosing an optimal spatial ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11235-3]
- 50. Comparison of imaging based single-cell resolution spatial ... [https://www.nature.com/articles/s41467-025-63414-1]
- 51. Single-cell transcriptomes identify patient-tailored ... [https://www.nature.com/articles/s41467-024-52980-5]
- 52. Omics Breakthroughs and the Future of Precision Medicine [https://www.psomagen.com/blog/omics-breakthroughs-precision-medicine]
- 53. Genomics and multiomics in the age of precision medicine [https://www.nature.com/articles/s41390-025-04021-0]
- 54. statistical learning methods for complex trait Comparing ... prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC11813155/]
- 55. Assessment of genomic prediction capabilities ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12423136/]
- 56. Miniaturized and Automatized Whole-Genome ... [https://www.protocols.io/view/miniaturized-and-automatized-whole-genome-amplific-cxgjxjun.html]
- 57. Single-cell RNA sequencing methods: plate-, droplet [https://www.integra-biosciences.com/united-states/en/blog/article/single-cell-rna-sequencing-methods-plate-droplet-and-microwell-based-approaches]
- 58. A Differential Depth Sequencing Method, SPRE‐Seq, for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12552348/]
- 59. A cost-effective sequencing method for genetic studies ... [https://www.nature.com/articles/s41525-024-00390-3]
- 60. Developing reproducible bioinformatics analysis workflows for... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2446-1]
- 61. Leveraging functional genomic annotations and ... [https://www.nature.com/articles/s41588-024-01704-y]
- 62. Feature Selection Stability and Accuracy of Prediction ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.611506/full]
- 63. EasyGeSe – a resource for benchmarking genomic prediction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12551357/]
- 64. A comparison of computational methods for expression ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03840-y]
- 65. Computational Strategies for Scalable Genomics Analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6947637/]
- 66. Systematic benchmarking of high-throughput subcellular ... [https://www.nature.com/articles/s41467-025-64292-3]
- 67. Systematic benchmarking of imaging spatial ... [https://www.nature.com/articles/s41467-025-64990-y]
- 68. 2025-2034 Cloud Genomics Market Outlook: Key Drivers, ... [https://www.openpr.com/news/4213794/2025-2034-cloud-genomics-market-outlook-key-drivers-emerging]
- 69. Scalable transcriptomics analysis with Dask - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9710082/]
- 70. AI-Driven Transcriptome Prediction in Human Pathology [https://pmc.ncbi.nlm.nih.gov/articles/PMC12189417/]
- 71. Comparing statistical learning methods for complex trait ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0317516]
- 72. Integrating gene expression data via weighted multiple kernel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12465700/]
- 73. A comparative study highlights superiority of LSTM in crop ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12540586/]
- 74. Gene selection for prediction of transcriptome signal based ... [https://link.springer.com/article/10.1007/s42452-025-07841-1]
- 75. Incorporating kernelized multi-omics data improves the ... [https://jasbsci.biomedcentral.com/articles/10.1186/s40104-022-00756-6]
- 76. New Study: Multi-Omics AI Model Boosts Accuracy to 90% ... [https://www.bgi.com/global/news/multi-omics-ai-model-boosts-accuracy-to-90-in-preterm-birth-prediction]
- 77. Improved genomic prediction performance with ensembles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12060242/]
- 78. comparing deep learning and GBLUP across diverse plant ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1568705/full]
- 79. Comparative analysis of regression algorithms for drug ... [https://bmcresnotes.biomedcentral.com/articles/10.1186/s13104-024-07026-w]
- 80. Personal transcriptome variation is poorly explained by ... [https://www.nature.com/articles/s41588-023-01574-w]
- 81. Resolving data bias improves generalization in binding ... [https://www.nature.com/articles/s42256-025-01124-5]
- 82. Predicting Drug Response and Synergy Using a Deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7737474/]
- 83. Establishing predictive machine learning models for drug ... [https://www.nature.com/articles/s41698-025-00937-2]
- 84. Prediction of protein–ligand binding affinity via deep learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10939342/]
- 85. Genomic selection in plant breeding: Key factors shaping ... [https://www.sciencedirect.com/science/article/pii/S1674205224000807]
- 86. Machine learning and related approaches in transcriptomics [https://www.sciencedirect.com/science/article/pii/S0006291X24007617]
- 87. Transcriptome-based prediction for polygenic traits in rice ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10803-3]
- 88. Comparing Genomic Prediction Models by Means of Cross ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.734512/full]
- 89. Cross-Validation Unleashed: The Key to Robust Model ... [https://medium.com/the-modern-scientist/cross-validation-unleashed-the-key-to-robust-model-performance-18441d52111d]