Gene Duplication as the Engine of NBS Gene Evolution: Mechanisms, Diversity, and Disease Resistance
This article provides a comprehensive analysis of gene duplication events in the evolution of Nucleotide-Binding Site-Leucine Rich Repeat (NBS-LRR) genes, the largest family of plant disease resistance (R) genes.
Gene Duplication as the Engine of NBS Gene Evolution: Mechanisms, Diversity, and Disease Resistance
Abstract
This article provides a comprehensive analysis of gene duplication events in the evolution of Nucleotide-Binding Site-Leucine Rich Repeat (NBS-LRR) genes, the largest family of plant disease resistance (R) genes. Aimed at researchers and scientists, we explore the foundational principles of NBS gene diversity, methodological approaches for identification and analysis, challenges in functional characterization, and validation techniques through comparative genomics and experimental assays. By synthesizing recent genomic studies across diverse plant species, we elucidate how duplication mechanisms—particularly tandem and whole-genome duplication—generate the genetic novelty essential for plant immunity, offering insights for future crop improvement and disease resistance breeding.
The Building Blocks of Immunity: Understanding NBS Gene Diversity and Duplication Mechanisms
The nucleotide-binding site-leucine-rich repeat (NBS-LRR) gene family represents one of the largest and most critical classes of disease resistance (R) genes in plants, serving as fundamental components of the plant immune system [1]. These genes encode intracellular receptor proteins that enable plants to detect pathogen invasions and initiate robust defense responses [2]. Since the cloning of the first NBS-LRR gene in 1994, extensive research has revealed their remarkable structural diversity and evolutionary dynamics [1]. These proteins function as specialized guards that monitor cellular homeostasis and trigger immune signaling upon perception of pathogen effectors [2]. The evolution of this gene family is characterized by frequent gene duplication events and subsequent functional diversification, making it a fascinating model for studying evolutionary genetics and host-pathogen co-evolution [3] [4]. This review provides a comprehensive overview of the NBS-LRR gene family, focusing on protein structure, classification, mechanisms in plant immunity, and evolutionary patterns driven by gene duplication.
Protein Structure and Classification
Domain Architecture
NBS-LRR proteins are large, multi-domain proteins typically ranging from 860 to 1,900 amino acids in length [1]. They share a conserved tripartite architecture consisting of:
- Variable N-terminal domain: Serves as a signaling platform and determines classification into major subfamilies.
- Central nucleotide-binding site (NBS) domain: Also known as the NB-ARC domain, contains several conserved motifs (P-loop, RNBS-A, RNBS-B, RNBS-C, GLPL, RNBS-D, MHD) that facilitate ATP/GTP binding and hydrolysis [1].
- C-terminal leucine-rich repeat (LRR) domain: Comprises multiple tandem LRR units that form a solenoid-shaped structure involved in specific protein-protein interactions and pathogen recognition [1].
The NBS domain functions as a molecular switch, where nucleotide-dependent conformational changes regulate signaling activity [1]. The LRR domain, with its extensive variation in sequence and repeat number, provides the structural basis for specific recognition of diverse pathogen effectors [2].
Classification System
Based on the identity of the N-terminal domain, NBS-LRR genes are primarily classified into three major subfamilies [3] [5]:
Table 1: Major Subfamilies of NBS-LRR Genes
| Subfamily | N-terminal Domain | Signaling Adaptors | Distribution | Representative Genes |
|---|---|---|---|---|
| TNL | Toll/Interleukin-1 Receptor (TIR) | EDS1, PAD4 [6] | Dicots only (absent in cereals) | RPS4 (Arabidopsis) [5] |
| CNL | Coiled-Coil (CC) | NRG1, ADR1 [6] | All angiosperms | RPM1, RPS2 (Arabidopsis) [2] |
| RNL | Resistance to Powdery Mildew 8 (RPW8) | ADR1 [6] | All angiosperms (reduced number) | RPH8A (Arabidopsis) [6] |
Additionally, based on domain combinations, the NBS-LRR family can be further divided into eight structural subtypes: CC-NBS (CN), CC-NBS-LRR (CNL), NBS (N), NBS-LRR (NL), RPW8-NBS (RN), RPW8-NBS-LRR (RNL), TIR-NBS (TN), and TIR-NBS-LRR (TNL) [7] [8].
NBS-LRR Genes in Plant Immunity
Effector-Triggered Immunity (ETI)
Plants employ a sophisticated two-layered immune system. The first layer, PAMP-triggered immunity (PTI), is activated by cell surface receptors recognizing conserved pathogen molecules [6]. Successful pathogens deliver effector proteins into plant cells to suppress PTI. As a countermeasure, the second layer, effector-triggered immunity (ETI), is mediated by NBS-LRR proteins that specifically recognize these effectors [6] [2]. ETI triggers a stronger, often localized defense response frequently accompanied by the hypersensitive response (HR), a form of programmed cell death at the infection site that restricts pathogen spread [9]. Recent studies indicate that PTI and ETI synergistically enhance plant immune responses [6].
Mechanisms of Pathogen Recognition
NBS-LRR proteins utilize distinct molecular strategies for pathogen detection, primarily through direct or indirect recognition.
Direct Recognition: Involves physical binding between the NBS-LRR protein and the pathogen effector. The LRR domain is typically responsible for this specific interaction.
Indirect Recognition (Guard Hypothesis): The NBS-LRR protein "guards" a host protein that is modified by a pathogen effector. The effector-induced modification of this host target is sensed by the NBS-LRR, activating defense.
- Examples: The Arabidopsis CNL protein RPM1 guards the host protein RIN4. The bacterial effectors AvrRpm1 and AvrB induce RIN4 phosphorylation, which is sensed by RPM1 [2]. Similarly, the Arabidopsis CNL RPS5 guards the kinase PBS1, detecting its cleavage by the bacterial protease effector AvrPphB [2].
The following diagram illustrates these two recognition models and the downstream signaling activation.
Downstream Signaling and Immune Activation
Recognition of a pathogen effector, whether direct or indirect, induces conformational changes in the NBS-LRR protein. This promotes the exchange of ADP for ATP in the NBS domain, transitioning the protein from an inactive to an active state [1] [2]. Activated NBS-LRR proteins oligomerize, forming resistosomes that initiate downstream signaling cascades [1]. Signaling pathways are often subfamily-specific:
- TNL signaling typically requires the lipase-like proteins EDS1 and PAD4, and often converges with the helper RNL protein ADR1 to activate defense gene expression [6].
- CNL signaling can engage helper RNLs like NRG1 and ADR1 to transduce immune signals [6].
This coordinated response leads to the activation of defense genes, production of antimicrobial compounds, and frequently the hypersensitive response.
Gene Duplication and Evolutionary Dynamics
Evolutionary Patterns Across Plant Lineages
Gene duplication is a primary driver of NBS-LRR gene family evolution, leading to significant variation in gene number across plant species [3] [4]. These duplications occur via whole-genome duplication (WGD) events, tandem duplication, and segmental duplication [4] [7]. The resulting copies can be retained through non-functionalization, neofunctionalization, or subfunctionalization, enabling plants to adapt to evolving pathogen populations.
Genome-wide studies reveal diverse evolutionary patterns:
- In Rosaceae, different species exhibit distinct patterns: Rosa chinensis shows "continuous expansion," while Fragaria vesca exhibits "expansion followed by contraction, then further expansion" [3] [5].
- In Solanaceae, potato shows "consistent expansion," tomato shows "expansion followed by contraction," and pepper shows "shrinking" [5].
- In Fabaceae, species like soybean and common bean exhibit a "consistently expanding" pattern [5].
Table 2: NBS-LRR Gene Counts and Evolutionary Patterns in Selected Plant Families
| Plant Family | Species | NBS-LRR Count | Primary Evolutionary Pattern | Key Duplication Mechanism |
|---|---|---|---|---|
| Rosaceae | Malus x domestica (Apple) | ~400 [4] | "Early sharp expansion to abrupt shrinking" [3] | Tandem & Segmental [3] |
| Rosaceae | Fragaria vesca (Strawberry) | 144 [9] | "Expansion, contraction, further expansion" [3] | Lineage-specific duplication [9] |
| Solanaceae | Nicotiana tabacum (Tobacco) | 603 [7] | Expansion (allotetraploid) | Whole-Genome Duplication [7] |
| Poaceae | Oryza sativa (Rice) | 508 [3] | "Contracting" [5] | Tandem [4] |
| Brassicaceae | Arabidopsis thaliana | ~150-207 [1] [6] | Moderate retention | Segmental & Tandem [4] |
Molecular Mechanisms of Duplication and Diversification
The birth-and-death evolution model explains the long-term dynamics of NBS-LRR genes, where duplicates are created and some are maintained while others are deleted or pseudogenized [1]. Key mechanisms include:
- Tandem Duplication: Leads to clusters of NBS-LRR genes that undergo sequence diversification through unequal crossing-over and gene conversion [1] [9].
- Whole-Genome Duplication: Provides raw genetic material for neofunctionalization, as observed in the allopolyploid formation of Nicotiana tabacum, where 76.62% of its NBS genes trace back to its parental genomes [7].
- Diversifying Selection: Acts predominantly on the LRR domain, particularly on solvent-exposed β-sheet residues, to generate variation for pathogen recognition [1].
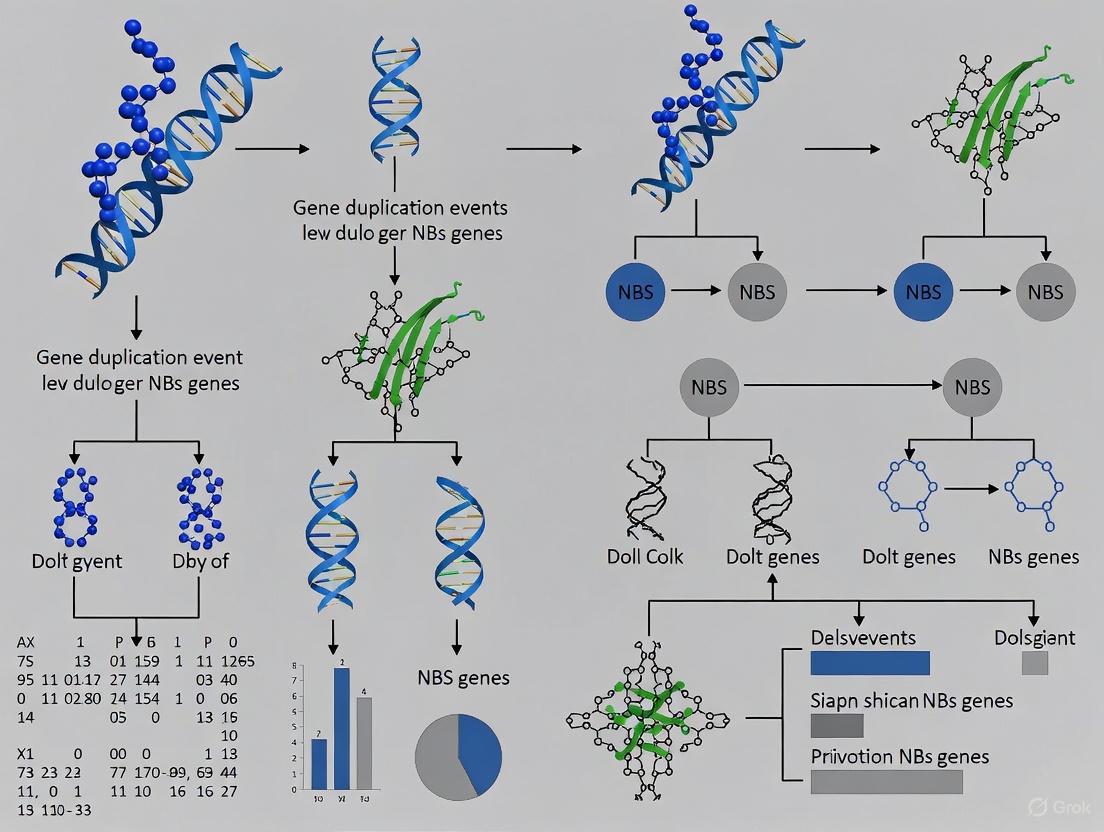
The following diagram summarizes the workflow for identifying NBS-LRR genes and analyzing their evolution, a common methodology in genomic studies.
Research Methods and Experimental Toolkit
Studying NBS-LRR genes requires a combination of bioinformatic and molecular biology techniques. Below is a standardized protocol for genome-wide identification and evolutionary analysis.
Standard Experimental Workflow
Step 1: Genome-Wide Identification
- Tool: HMMER v3.1b2 with NB-ARC domain (PF00931) HMM profile.
- Parameters: Default settings, E-value threshold (e.g., 1.0 or 10⁻⁴) [3] [9].
- Validation: Confirm candidate genes using Pfam and NCBI-CDD for NBS, TIR (PF01582), CC (PF18052), RPW8 (PF05659), and LRR domains [3] [7].
Step 2: Phylogenetic and Evolutionary Analysis
- Sequence Alignment: Use MUSCLE or ClustalW for multiple sequence alignment of NBS domains or full-length proteins [7] [9].
- Tree Construction: Build phylogenetic trees using Maximum Likelihood (e.g., MEGA11, FastTree) with 1000 bootstrap replicates [7] [9].
- Duplication Analysis: Use MCScanX to identify tandem and segmental duplications. Calculate non-synonymous (Ka) and synonymous (Ks) substitution rates with KaKs_Calculator to infer selective pressure [7] [9].
Step 3: Expression Profiling
- Data Source: Process RNA-seq data from public repositories (e.g., NCBI SRA).
- Method: Map reads to reference genome using Hisat2, perform transcript quantification with Cufflinks/Cuffdiff, and identify differentially expressed genes (DEGs) [7].
Essential Research Reagents and Tools
Table 3: Key Reagents and Tools for NBS-LRR Gene Research
| Category | Reagent/Tool | Specific Example/Function | Application in Research |
|---|---|---|---|
| Bioinformatic Tools | HMMER | PF00931 (NB-ARC) Hidden Markov Model | Identify NBS-domain containing genes [3] [7] |
| Bioinformatic Tools | MCScanX | Collinearity detection algorithm | Identify segmental and tandem gene duplications [7] |
| Bioinformatic Tools | KaKs_Calculator | NG (Nei-Gojobori) model | Calculate Ka/Ks ratio to assess selective pressure [7] |
| Molecular Biology | RNA-seq Libraries | SRA accessions (e.g., SRP141439) | Profile NBS-LRR gene expression during infection [7] |
| Molecular Biology | ClustalW/MUSCLE | Multiple sequence alignment | Prepare data for phylogenetic analysis [7] [9] |
The NBS-LRR gene family stands as a cornerstone of plant immunity, enabling specific pathogen recognition through direct and indirect mechanisms. Its evolutionary trajectory is profoundly shaped by gene duplication events, including whole-genome, tandem, and segmental duplications, followed by functional diversification via birth-and-death evolution. This dynamic process creates a vast repertoire of receptors, allowing plants to adapt to rapidly evolving pathogens. Future research leveraging expanding genomic resources and functional genomic tools will continue to unravel the intricate mechanisms of NBS-LRR function and evolution, providing insights crucial for engineering durable disease resistance in crops.
Gene duplication is a fundamental mechanism for evolutionary innovation, generating genetic raw material for new functions and complex traits. Two primary processes, Whole-Genome Duplication (WGD) and Tandem Duplication (TD), have shaped the genomes of eukaryotes, particularly plants, through dramatically different mechanisms and evolutionary consequences [4]. Understanding the distinct roles of these duplication types is especially crucial for research on Nucleotide Binding Site (NBS) gene evolution, as these disease resistance genes exhibit distinctive patterns of retention and divergence following different duplication events [10] [11]. This review provides a comprehensive technical analysis of how WGD and TD serve as major expansion drivers, their differential impacts on gene fate, and methodologies for their study, with specific application to NBS gene research.
Mechanisms and Prevalence of Duplication Types
Whole-Genome Duplication (WGD)
WGD, or polyploidization, represents the most extensive form of gene duplication, creating a sudden duplication of the entire gene set and increasing genome size instantaneously [4]. Unlike most other eukaryotes, plant genomes have experienced recurrent WGD events throughout their evolutionary history, with these events occurring multiple times over the past 200 million years of angiosperm evolution [4]. Following WGD, the polyploid genome undergoes a process of "fractionation," where chromosomal rearrangements, gene conversions, heightened transposon activity, and epigenetic changes lead to a reduced set of duplicate gene pairs over evolutionary time [10]. The prevalence of WGD is demonstrated by the fact that on average, 65% of annotated genes in plant genomes have a duplicate copy, with most derived from WGD events [4].
Tandem Duplication (TD)
In contrast to WGD, tandem duplication involves the localized amplification of specific genomic regions, typically through unequal recombination between interspersed repetitive elements during meiosis or recombinational repair [10]. This process results in the creation of clusters of duplicated genes that are adjacent to each other on the chromosome. TD can also occur through insertion of retrotransposed genes, though these often lack promoters and are frequently pseudogenized at birth [10]. Tandemly duplicated regions are genetically unstable and can be readily lost or amplified further by recombination, with stability highly correlated with segment length [12]. The spontaneous mutation rate for tandem duplications is high, with approximately 10% of bacteria in growing cultures containing gene duplications somewhere in the genome [12].
Table 1: Fundamental Characteristics of WGD and TD
| Characteristic | Whole-Genome Duplication (WGD) | Tandem Duplication (TD) |
|---|---|---|
| Genomic Scale | Entire genome | Focal regions (kb to Mb) |
| Mechanism | Polyploidization | Unequal recombination, replication slippage |
| Frequency in Plants | Recurrent throughout evolution | Continuous, spontaneous |
| Stability | Stable after diploidization | Highly unstable, length-dependent |
| Typical Gene Copy Number | 2 (initially) | Variable (2 to 15+) |
| Prevalence | ~65% of plant genes have WGD-derived paralogs | ~10% of human genome consists of TDs |
Differential Retention and Functional Bias
Empirical evidence demonstrates that WGD and TD exhibit striking differences in the functional categories of genes they preserve, reflecting their distinct evolutionary roles and selective constraints.
Retention Patterns and Functional Specialization
Comparative analysis in Populus trichocarpa reveals that WGD and TD retain fundamentally different gene sets. WGD-derived duplicates are significantly longer (700 bp longer on average), expressed in more tissues (20% greater expression breadth), and enriched for transcription factors, signal transduction components, and DNA-binding proteins [10]. This pattern aligns with the gene balance hypothesis, which predicts that dosage-sensitive genes involved in macromolecular complexes and regulatory networks are preferentially retained after WGD to maintain stoichiometric balance [10] [4].
Conversely, TD genes are significantly shorter, exhibit more tissue-specific expression, and are overwhelmingly enriched for environmental interaction genes, particularly disease resistance genes (NBS-LRRs), receptor-like kinases (RLKs), and stress-responsive genes [10] [11] [13]. This functional bias creates a "core-adaptive" model of gene evolution, where different duplication mechanisms maintain distinct functional genomic compartments [11].
Evolutionary Implications for NBS Genes
The concentration of NBS-LRR genes in tandem arrays represents a key adaptation for evolutionary arms races against rapidly evolving pathogens [10] [11] [14]. TD provides a mechanism for rapid generation of genetic diversity through recurrent duplication and birth-death evolution, creating variation in pathogen recognition specificities [14]. Studies of maize ZmNBS genes reveal extensive presence-absence variation, distinguishing conserved "core" subgroups from highly variable "adaptive" subgroups, with tandem and proximal duplications showing signs of relaxed or positive selection compared to the strong purifying selection on WGD-derived duplicates [11].
Table 2: Functional and Evolutionary Properties of Retained Duplicates
| Property | WGD-Derived Genes | TD-Derived Genes |
|---|---|---|
| Preferred Functional Categories | Transcription factors, Signal transduction components | Disease resistance (NBS-LRR), Receptor-like kinases, Stress response |
| Selection Pressure | Strong purifying selection (Low Ka/Ks) | Relaxed or positive selection |
| Expression Profile | Broad expression (20% more tissues) | Tissue-specific expression |
| Structural Features | Longer genes (700 bp longer) | Shorter genes |
| Role in Evolution | Conservation of core regulatory networks | Rapid adaptation, arms races |
| Genetic Diversity | Lower diversity, purifying selection | High diversity, positive selection |
Evolutionary Dynamics and Functional Divergence
Divergence Mechanisms and Trajectories
Following duplication, genes may evolve through several trajectories: retention of original function (functional conservation), partitioning of ancestral functions (subfunctionalization), acquisition of novel functions (neofunctionalization), or degradation into nonfunctional pseudogenes (nonfunctionalization) [10] [4]. The distribution of expression divergence for WGD-derived pairs in Populus suggests nearly half have diverged by a random degenerative process, while the remaining pairs exhibit more conserved expression than expected by chance, consistent with selective constraints of gene balance [10].
The duplication-degeneration-complementation (DDC) model proposes that degenerative mutations in regulatory elements can preserve duplicates by making both copies necessary to maintain the full complement of ancestral functions [10]. This process may work in concert with neofunctionalization, as degenerative processes affecting silencer elements could potentially promote the acquisition of new expression patterns [10].
Impact on Structural Variants and Genetic Load
Recent research in Cochlearia autopolyploids reveals complex interactions between WGD and structural variant (SV) evolution. WGD increases the masking of recessive deleterious mutations, leading to progressive accumulation of deleterious SVs across ploidal levels (diploids to octoploids), potentially reducing adaptive potential [15]. However, polyploids also exhibit more ploidy-specific SVs with signals of local adaptation, suggesting SV accumulation may provide benefits alongside costs [15]. This dual impact creates contrasting evolutionary dynamics where SVs simultaneously contribute to genetic load while potentially providing raw material for adaptation.
Methodological Approaches for Detection and Analysis
Experimental Protocols for Duplication Analysis
WGD Identification Protocol:
- Synteny Analysis: Identify collinear genomic blocks between related species or within genomes using tools like MCScanX
- Ks Distribution Analysis: Calculate synonymous substitution rates (Ks) for paralogous pairs; WGD events appear as peaks in Ks distributions
- Phylogenomic Dating: Reconstruct gene trees for multigene families and reconcile with species trees to identify duplication events
- Cytogenetic Confirmation: Chromosome counting and karyotype analysis to validate ploidy level
TD Detection Protocol (DTDHM Methodology) [16]:
- Read Processing: Convert NGS data to BAM files after quality control and alignment
- Signal Extraction: Calculate Read Depth (RD) and Mapping Quality (MQ) signals across the genome using sliding windows
- Signal Smoothing: Apply Total Variation (TV) model to reduce noise in RD and MQ signals
- Segmentation: Use Circular Binary Segmentation (CBS) algorithm to identify candidate variant regions
- Classification: Apply K-nearest neighbor (KNN) algorithm with RD and MQ as features to predict TD regions
- Boundary Refinement: Extract split reads and discordant read pairs to precisely define duplication boundaries
Computational Framework for Duplication Analysis
The following workflow illustrates the integrated approach for detecting and analyzing tandem duplications from NGS data:
Figure 1: DTDHM Workflow for Tandem Duplication Detection
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Duplication Studies
| Reagent/Resource | Function/Application | Example Use |
|---|---|---|
| Lambda Red Recombinase System | Facilitates homologous recombination for engineered duplications | Constructing defined duplications in bacterial systems [12] |
| Oxford Nanopore/PacBio | Long-read sequencing for SV detection | Resolving complex duplicated regions [15] |
| Sniffles2 | SV caller for long-read data | Identifying SVs in autopolyploid samples [15] |
| DTDHM Pipeline | TD detection from short-read data | Comprehensive TD identification in human genomes [16] |
| Droplet Digital PCR (ddPCR) | Absolute copy number quantification | Validating duplication structure and copy number [12] |
| MorexV3 Barley Genome | High-quality reference genome | Studying association between arms-race genes and LDPRs [14] |
Implications for NBS Gene Evolution Research
The differential impact of WGD and TD has profound implications for understanding NBS gene evolution. The concentration of NBS genes in tandem arrays reflects an evolutionary strategy to generate diversity for pathogen recognition [11] [14]. Lineages where NBS genes are physically associated with duplication-prone genomic regions enjoy selective advantages in host-pathogen arms races [14].
Analysis of ZmNBS genes in maize reveals that duplication mechanisms significantly impact evolutionary rates: WGD-derived genes exhibit strong purifying selection, while TD-derived genes show signs of relaxed or positive selection [11]. This pattern supports the hypothesis that TD provides a substrate for rapid adaptation in resistance genes. Furthermore, presence-absence variation distinguishes conserved "core" NBS subgroups from highly variable "adaptive" subgroups, creating a dynamic evolutionary landscape [11].
Recent findings in barley demonstrate that natural selection has favored lineages where pathogen defense genes are associated with duplication-inducing sequences, particularly kilobase-scale tandem repeats [14]. This association between "arms-race genes" and duplication-inducing elements represents an effective cooperative relationship at the genomic level, facilitating rapid adaptation to evolving pathogen threats.
WGD and TD serve as complementary drivers of genomic expansion with distinct evolutionary impacts. WGD preferentially preserves dose-sensitive regulatory genes through strong purifying selection, maintaining stoichiometric balance in core cellular processes. In contrast, TD rapidly generates diversity for environmental interaction genes, particularly NBS-type disease resistance genes, through recurrent duplication and birth-death evolution. The integration of advanced detection methodologies, from long-read sequencing to hybrid computational approaches, enables comprehensive characterization of these duplication processes. For NBS gene research, understanding these differential duplication mechanisms provides critical insights into the evolutionary dynamics of disease resistance and adaptive potential in plants, with significant implications for crop improvement and sustainable agriculture. Future research should focus on integrating multi-omics data to precisely trace the evolutionary trajectories of duplicated genes and their contributions to adaptive phenotypes.
The NBS-LRR gene family constitutes one of the most critical components of the plant immune system, encoding intracellular receptors that recognize pathogen effectors and trigger defense responses. The genomic organization of these genes is not random; they frequently exhibit cluster arrangements and uneven distribution across chromosomes, patterns shaped by extensive gene duplication events. These duplication events, including tandem duplications and segmental duplications, provide raw genetic material for evolutionary innovation, enabling plants to rapidly adapt to evolving pathogen pressures.
Understanding the principles governing NBS gene distribution and the mechanisms driving their expansion is crucial for deciphering plant-pathogen co-evolution and for developing novel crop improvement strategies. This review synthesizes recent genome-wide studies across diverse plant species to elucidate common patterns and unique features of NBS gene genomic architecture, with particular emphasis on the role of gene duplication in their evolution.
Methodology for Genome-Wide Identification of NBS Genes
Standard Bioinformatics Pipeline
The accurate identification and classification of NBS-LRR genes across plant genomes relies on a standardized bioinformatics approach that leverages conserved protein domains. The typical workflow integrates multiple computational tools to ensure comprehensive gene discovery and annotation [17] [18].
Table 1: Key Bioinformatics Tools for NBS Gene Identification
| Tool Category | Specific Tool | Purpose | Key Parameters |
|---|---|---|---|
| Domain Search | HMMER | Identify NB-ARC domains (PF00931) | E-value threshold (1e-20) [19] |
| Domain Verification | Pfam/NCBI CDD | Confirm additional domains (TIR, CC, LRR) | Domain architecture analysis [20] |
| Multiple Sequence Alignment | MUSCLE/Mafft | Align protein sequences for phylogenetic analysis | Default parameters [17] |
| Phylogenetic Analysis | MEGA11 | Construct evolutionary trees | Maximum likelihood, 1000 bootstraps [17] [19] |
| Duplication Analysis | MCScanX | Identify segmental and tandem duplications | BLASTP followed by collinearity detection [17] |
The process typically begins with HMMER searches using the NB-ARC domain model (PF00931) from the Pfam database against the proteome of the target species [17] [18]. Candidate genes are then verified through domain architecture analysis using resources like the NCBI Conserved Domain Database to classify genes into subfamilies based on their N-terminal domains (TIR, CC, or RPW8) and C-terminal LRR regions [17]. This classification enables researchers to categorize NBS genes into major subfamilies: TNL (TIR-NBS-LRR), CNL (CC-NBS-LRR), RNL (RPW8-NBS-LRR), and various partial domains [18].
Experimental Validation Approaches
While bioinformatics predictions provide comprehensive datasets, experimental validation remains crucial for confirming gene models and expression patterns. Common experimental approaches include:
- RNA-seq analysis: Mapping transcriptomic data to identified NBS genes to verify expression and alternative splicing patterns [17].
- qRT-PCR: Quantitative validation of candidate gene expression under pathogen challenge or specific stress conditions [18].
- Virus-Induced Gene Silencing (VIGS): Functional characterization of specific NBS genes through targeted silencing and subsequent phenotyping [20].
Figure 1: Workflow for comprehensive identification and validation of NBS genes, integrating bioinformatics and experimental approaches
Comparative Genomic Distribution Across Plant Species
Chromosomal Distribution Patterns
NBS-LRR genes consistently display non-random distribution patterns across plant genomes, with significant variations in gene counts and densities across chromosomes. Recent multi-species analyses reveal both conserved and species-specific distribution characteristics.
Table 2: Comparative Genomic Distribution of NBS Genes Across Plant Species
| Plant Species | Total NBS Genes | Chromosomal Range | Distribution Hotspots | Clustered Genes |
|---|---|---|---|---|
| Capsicum annuum (Pepper) | 252 | All 12 chromosomes + unassigned | Chromosome 3 (38 genes) | 54% (136 genes in 47 clusters) [21] |
| Raphanus sativus (Radish) | 225 | 9 chromosomes + scaffolds | U blocks (R02, R04, R08) | 72% in 48 clusters [18] |
| Nicotiana tabacum (Tobacco) | 603 | Parental genome contributions | N/A | Significant tandem duplication [17] |
| Solanum tuberosum (Potato) | 587 domains | 12 chromosomes | Multiple clusters | Stacked arrangement with complete/incomplete genes [22] |
| Gossypium hirsutum (Cotton) | 12,820 (across 34 species) | Wide variation | Species-specific | 168 domain architecture classes [20] |
In pepper (Capsicum annuum), comprehensive analysis identified 252 NBS-LRR genes distributed across all chromosomes, with chromosome 3 harboring the highest concentration (38 genes) while chromosomes 2 and 6 contained the lowest (5 genes each) [21]. Similarly, in radish (Raphanus sativus), researchers identified 225 NBS-encoding genes with 202 mapped to chromosomes and 23 on scaffolds, showing uneven distribution across the genome with concentration in specific chromosomal blocks [18].
A remarkable case of NBS gene expansion is observed in tobacco (Nicotiana tabacum), an allotetraploid formed from hybridization of N. sylvestris and N. tomentosiformis. The 603 NBS genes identified in N. tabacum represent approximately the combined total of its parental species (344 and 279 respectively), with 76.62% of these genes traceable to their parental genomes, demonstrating the impact of polyploidization on NBS gene repertoire expansion [17].
Physical Clustering of NBS Genes
A predominant feature of NBS gene genomic organization is their tendency to form physical clusters. These clusters, primarily driven by tandem duplication events, represent hotspots for genetic innovation and functional diversification.
In pepper, 54% of NBS-LRR genes (136 genes) are organized into 47 distinct physical clusters distributed across the genome, with chromosome 3 containing the highest number of clusters (10) and the largest single cluster comprising 8 genes [21]. Cluster composition varies, with some containing members exclusively from the same gene subfamily while others exhibit mixing of different subfamilies, reflecting complex evolutionary histories.
Similarly, in radish, a substantial majority (72%) of NBS-encoding genes are grouped in 48 clusters distributed in 24 crucifer blocks, with the U block on chromosomes R02, R04, and R08 containing the highest concentration (48 genes) [18]. These clusters were found to be predominantly homogeneous, containing NBS-encoding genes derived from recent common ancestors, suggesting recent expansion events.
The potato genome exhibits a particularly clustered organization, with NBS-LRR genes occurring in stacked arrangements where complete, potentially functional genes alternate with incomplete ones. This organization is believed to serve as a reservoir for variation, enabling the production of new functional R alleles through frameshift recombination and DNA repair processes [22].
Gene Duplication Mechanisms in NBS Gene Evolution
Duplication Types and Rates
Gene duplication plays a fundamental role in the expansion and evolution of NBS gene families, with different mechanistic pathways contributing to their diversification across plant lineages.
Whole-genome duplication (WGD) events have significantly contributed to NBS gene expansion in several species. In tobacco, analysis of the allotetraploid genome revealed that WGD contributed substantially to the expansion of NBS gene families, with the tobacco genome containing approximately the combined NBS gene count of its diploid progenitors [17]. Similarly, in cotton, analyses revealed that segmental and whole-genome duplications were the primary drivers of EDS1 gene family expansion, a key component in NBS-mediated signaling [19].
Tandem duplication represents another major mechanism for NBS gene expansion. In radish, researchers identified 15 tandem duplication events and 20 segmental duplication events in the NBS family, highlighting the importance of both small-scale and large-scale duplication mechanisms [18]. These duplication events create genetic redundancy that allows for functional diversification through neofunctionalization or subfunctionalization.
Evolutionary Dynamics of Duplicated Genes
Following duplication events, NBS genes undergo different evolutionary fates shaped by natural selection. Analysis of selection pressures typically involves calculating non-synonymous (Ka) and synonymous (Ks) substitution rates, with Ka/Ks ratios indicating the mode of selection.
In cotton EDS1 genes, Ka/Ks analysis revealed that most duplicates were under purifying selection (Ka/Ks < 1), indicating selective constraint and functional conservation [19]. Similarly, comparative analysis of NBS genes across 34 land plant species identified both core orthogroups (conserved across species) and unique orthogroups (species-specific), reflecting varying evolutionary trajectories [20].
The concept of "birth-and-death" evolution is particularly relevant for NBS genes, whereby new genes are created by duplication while others are inactivated or deleted through pseudogenization. This dynamic process generates considerable interspecific and intraspecific variation in NBS gene content and organization, contributing to the evolutionary arms race between plants and their pathogens [23].
Figure 2: Gene duplication mechanisms and their evolutionary consequences in NBS gene family expansion
Functional Implications of Genomic Distribution Patterns
Association with Disease Resistance Loci
The non-random distribution of NBS genes has significant functional implications, particularly in their association with known disease resistance loci. Studies across multiple Brassica species have demonstrated that certain classes of resistance genes, particularly receptor-like kinases (RLKs) and receptor-like proteins (RLPs), are frequently co-localized with reported disease resistance loci [24]. This spatial association suggests that genomic context influences resistance gene function and evolution.
Phylogenetic analysis of cloned R genes and QTL-mapped RLKs and RLPs has identified distinct clusters, enhancing our understanding of their evolutionary trajectories and functional relationships [24]. These analyses reveal that NBS genes with similar genomic distributions often share evolutionary histories and potentially related functions.
Expression Variation and Regulatory Mechanisms
The genomic distribution of NBS genes influences their expression patterns and regulatory mechanisms. Expression profiling of radish NBS genes identified 75 NBS-encoding genes that contributed to resistance against Fusarium wilt, with differential expression patterns between resistant and susceptible varieties [18]. Detailed analysis revealed that RsTNL03 (Rs093020) and RsTNL09 (Rs042580) expression positively regulated radish resistance to Fusarium oxysporum, while RsTNL06 (Rs053740) expression functioned as a negative regulator [18].
Similarly, comprehensive transcriptomic analysis of NBS genes across multiple species identified distinct expression patterns, with orthogroups OG2, OG6, and OG15 showing putative upregulation in different tissues under various biotic and abiotic stresses in cotton accessions with contrasting susceptibility to cotton leaf curl disease [20]. These expression differences highlight the functional significance of NBS gene distribution and duplication events.
Association with Duplication-Prone Genomic Regions
Emerging evidence suggests that NBS genes are frequently associated with duplication-prone genomic regions, creating a evolutionary advantage in pathogen-host arms races. Research in barley has demonstrated that natural selection has favored lineages in which arms-race genes—particularly pathogen defense genes—are associated with duplication-inducers, most notably Kb-scale tandem repeats [25].
Such duplication-prone regions show a history of repeated long-distance 'dispersal' to distant genomic sites, followed by local expansion by tandem duplication. This association between duplication-inducing elements and NBS genes creates effectively cooperative associations that enhance the generation of genetic diversity, providing raw material for evolutionary innovation in pathogen recognition [25].
Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for NBS Gene Studies
| Reagent/Resource | Specific Example | Application | Reference |
|---|---|---|---|
| Primer Sets | P-loop, Kinase-2, GLPL primers | NBS domain amplification and profiling | [22] |
| HMM Profiles | PF00931 (NB-ARC) | Domain identification and gene annotation | [17] [18] |
| Genome Databases | CottonMD, Phytozome, NCBI | Genomic sequence retrieval | [19] [20] |
| Software Tools | MCScanX, OrthoFinder, MEGA11 | Evolutionary and duplication analysis | [17] [20] |
| Expression Databases | IPF database, CottonFGD | Expression pattern analysis | [20] |
| VIGS Vectors | Tobacco rattle virus-based systems | Functional validation of candidate genes | [20] |
The genomic distribution of NBS genes exhibits conserved patterns across plant species, characterized by physical clustering and uneven chromosomal distribution. These patterns are primarily driven by various duplication mechanisms, including tandem duplication, segmental duplication, and whole-genome duplication, which collectively expand and diversify the NBS gene repertoire. The concentration of NBS genes in duplication-prone genomic regions facilitates rapid evolution of pathogen recognition capabilities, directly supporting the "arms race" model of plant-pathogen co-evolution.
Understanding these distribution patterns and their evolutionary origins has significant practical implications for crop improvement strategies. The association between specific NBS gene clusters and disease resistance phenotypes enables more efficient marker-assisted selection and targeted breeding approaches. Furthermore, characterizing the duplication mechanisms that shape NBS gene evolution provides insights for developing synthetic biology approaches to enhance disease resistance in crop plants. Future research integrating pan-genomic analyses with functional studies will further elucidate the complex relationship between genomic distribution, evolutionary history, and disease resistance function in NBS genes.
This whitepaper examines the pervasive pattern of TIR-NBS-LRR (TNL) gene loss in monocot lineages, a compelling model of lineage-specific evolution within the nucleotide-binding site-leucine-rich repeat (NBS-LRR) gene family. Plant NBS-LRR genes, the largest category of disease resistance (R) genes, are crucial intracellular immune receptors that mediate effector-triggered immunity (ETI). While TNL genes are prevalent in dicots, they are conspicuously absent in most monocot genomes. Recent research leveraging chromosome-level genomes and synteny analysis has revealed that this gene loss pattern originated from a specific genomic deletion event in a common monocot ancestor, followed by subsequent diversification of remaining NLR classes. This evolutionary trajectory exemplifies how gene duplication, domain degeneration, and selection pressures collectively shape genomic architecture and functional diversity in plant immune systems across lineages.
NBS-LRR Gene Structure and Classification
NBS-LRR genes encode a pivotal class of plant immune receptors responsible for recognizing pathogen effectors and initiating robust defense responses [26]. These proteins typically consist of three core domains:
- N-terminal domain: Provides signaling specificity and falls into three major types - TIR (Toll/Interleukin-1 Receptor), CC (Coiled-Coil), or RPW8 (Resistance to Powdery Mildew 8)
- NB-ARC (Nucleotide-Binding Adaptor Shared by APAF-1, R proteins, and CED-4) domain: Binds and hydrolyzes ATP/GTP, functioning as a molecular switch for activation
- LRR (Leucine-Rich Repeat) domain: Mediates protein-protein interactions and pathogen recognition through sequence hypervariability
Based on their N-terminal domains, NBS-LRR genes are classified into three principal subfamilies: TNL (TIR-NBS-LRR), CNL (CC-NBS-LRR), and RNL (RPW8-NBS-LRR) [26]. The RNL subfamily is further divided into NRG1 (N-required gene 1) and ADR1 (Activated disease resistance gene 1) lineages [26]. This structural classification reflects functional specialization within plant immune networks, with TNL and CNL proteins primarily responsible for pathogen recognition, while RNL proteins often function in downstream defense signal transduction.
Evolutionary Dynamics of NBS-LRR Genes
The NBS-LRR gene family exhibits remarkable evolutionary dynamism, characterized by several distinctive features:
- Rampant gene duplication and loss: Creating substantial variation in gene copy number across species [27]
- High sequence diversity: Particularly in LRR domains that interact directly with pathogen effectors [28]
- Frequent domain rearrangements: Including domain loss, fusion, and the emergence of chimeric proteins [28]
- Lineage-specific evolutionary patterns: Resulting from differential selection pressures across plant taxa
These characteristics make the NBS-LRR gene family an exemplary system for studying lineage-specific evolution. The disproportionate loss of TNL genes in monocots represents one of the most striking examples of such lineage-specific patterns, with profound implications for understanding the evolutionary malleability of plant immune systems.
The Monocot TNL Loss Phenomenon: Patterns and Evidence
Distribution of NBS-LRR Subfamilies Across Angiosperms
Comparative genomic analyses across multiple plant species have revealed a consistent pattern of TNL absence in monocot lineages. The table below summarizes the distribution of NBS-LRR subfamilies across representative plant species:
Table 1: NBS-LRR Gene Distribution Across Plant Species
| Species | Classification | CNL Genes | TNL Genes | RNL Genes | Total NBS-LRR | Reference |
|---|---|---|---|---|---|---|
| Arabidopsis thaliana | Eudicot | 40 | 48 | 18 | 106 | [29] |
| Dendrobium officinale | Monocot (Orchid) | 10 | 0 | 9 | 19 | [29] |
| Dendrobium nobile | Monocot (Orchid) | 18 | 0 | 14 | 32 | [29] |
| Dendrobium chrysotoxum | Monocot (Orchid) | 14 | 0 | 9 | 23 | [29] |
| Arachis hypogaea cv. Tifrunner | Eudicot (Peanut) | 118 | 229 | Not specified | 713 | [28] |
| Akebia trifoliata | Eudicot | 50 | 19 | 4 | 73 | [26] |
| Vanilla planifolia | Monocot (Orchid) | 2 | 0 | 2 | 4 | [29] |
| Apostasia shenzhenica | Monocot (Orchid) | 4 | 0 | 3 | 7 | [29] |
The comprehensive absence of TNL genes in monocot species, contrasted with their consistent presence in eudicots, indicates this gene loss occurred early in monocot evolution, prior to the diversification of major monocot lineages.
Genomic Evidence for TNL Loss in Monocots
Recent synteny-informed phylogenetic analyses provide compelling evidence for the mechanism underlying TNL loss in monocots. A 2025 study introduced a refined classification system for angiosperm NLR genes that categorizes them into five distinct classes: CNLA, CNLB, CNL_C, TNL, and RNL [27]. This classification revealed:
- Microsynteny correspondence: Clear syntenic relationships exist between non-TNL genes in monocots and the extinct TNL subclass in ancestral species
- Genomic deletion event: The TNL loss in monocots likely resulted from a specific genomic deletion in a common monocot ancestor, rather than independent gene losses across multiple lineages
- Subfunctionalization of remaining classes: Following TNL loss, the remaining CNL subclasses expanded and diversified to compensate functionally
This synteny-based evidence suggests that the extinction of TNL genes in monocots was not a gradual process but rather a discrete genomic event that shaped subsequent immune system evolution in monocot lineages.
Evolutionary Mechanisms Driving TNL Loss and Genomic Diversification
Genomic and Evolutionary Processes
Several interconnected evolutionary mechanisms have contributed to the lineage-specific patterns of NBS-LRR gene evolution, including TNL loss in monocots:
Lineage-Specific Genomic Deletion The initial TNL loss in monocots likely resulted from a significant genomic deletion event affecting a chromosomal region housing multiple TNL genes [27]. This event potentially created selective pressures favoring the expansion and diversification of remaining CNL classes to compensate for the lost TNL functions.
Domain Degeneration and Gene Structure Variation NBS genes frequently undergo structural variations, including:
- LRR domain loss: Creating truncated NBS proteins that may retain signaling functions [29]
- NB-ARC domain degeneration: Reducing functionality and potentially leading to pseudogenization [29]
- Domain fusion events: Creating novel chimeric proteins, such as NBS-WRKY fusions observed in peanut genomes [28]
Differential Selection Pressures Evolutionary analyses reveal distinct selection patterns acting on different NBS-LRR components:
- LRR domains: Experience more relaxed selection or positive selection due to direct pathogen effector interactions [28]
- NBS domains: Generally under stronger purifying selection due to conserved ATP/GTP binding functions
- N-terminal domains: Show lineage-specific selection patterns corresponding to functional specialization
Gene Duplication as a Driver of Lineage-Specific Evolution
Gene duplication events play a central role in NBS-LRR gene evolution, with different duplication mechanisms contributing to genomic diversification:
Table 2: Gene Duplication Mechanisms in NBS-LRR Evolution
| Duplication Mechanism | Characteristics | Evolutionary Impact | Examples |
|---|---|---|---|
| Tandem Duplication | Clustered gene arrays on chromosomes; Rapid expansion of specific gene families | Generates genetic material for neofunctionalization; Creates resistance gene clusters | Primary mechanism in Akebia trifoliata (33 genes) [26] |
| Dispersed Duplication | Non-clustered distribution throughout genome; May involve transposition elements | Enables subfunctionalization; Allows genomic repositioning | Significant contributor in Akebia trifoliata (29 genes) [26] |
| Whole Genome Duplication | Polyploidization events; Affects entire genomic complement | Provides raw material for specialization; Can lead to fractionation | Observed in Arachis hypogaea (allotetraploid) [28] |
| Segment Duplication | Duplication of chromosomal segments; Contains multiple genes | Preserves gene neighborhoods; Maintains regulatory contexts | Inferred from synteny analyses [27] |
These duplication mechanisms interact with lineage-specific evolutionary pressures to shape the NLR gene repertoire in different plant species. In monocots, following TNL loss, duplication of remaining CNL classes appears to have been a crucial compensatory mechanism for maintaining immune system functionality.
Experimental Approaches for Studying Lineage-Specific NBS-LRR Evolution
Genomic Identification and Classification Protocols
Comprehensive NBS Gene Identification Pipeline
- Initial Sequence Retrieval
- Obtain genome assemblies and annotation files from databases (NCBI, Phytozome)
- Extract all protein-coding sequences for analysis
Domain-Based Identification
- Perform HMMER searches using NB-ARC domain profile (PF00931) with E-value cutoff of 1.0 [26]
- Conduct BLASTP analyses against known NBS proteins using NB-ARC domain as query
- Merge candidate lists and remove redundancies
Domain Verification and Classification
Manual Curation
- Verify domain organization and gene structure
- Remove pseudogenes and partial sequences
- Confirm presence of intact NBS domains for evolutionary analyses
Synteny-Informed Classification Methodology Recent advances incorporate microsynteny analysis for improved NLR classification [27]:
- Microsynteny Network Construction: Identify conserved gene neighborhoods across species
- Synteny-Based Classification: Categorize NLR genes into CNLA, CNLB, CNL_C, TNL, and RNL classes
- Evolutionary Reconciliation: Map gene relationships using synteny conservation rather than sequence similarity alone
Evolutionary Analysis Methods
Selection Pressure Analysis
- Gene Family Alignment
- Generate multiple sequence alignments using MAFFT or MUSCLE
- Filter alignments for quality (≤10% gaps, ≥80% identity in non-gapped positions)
Phylogenetic Reconstruction
- Construct gene trees using maximum likelihood methods (RAxML, IQ-TREE)
- Reconcile gene trees with species tree using tools like SoftParsMap [30]
Selection Detection
- Calculate nonsynonymous/synonymous substitution rates (dN/dS) using branch models
- Identify lineages with dN/dS >1 indicating positive selection
- Account for dS saturation by excluding comparisons with dS >3 [30]
Duplicate Gene Analysis
- Duplication Event Identification
- Map gene locations to chromosomes
- Identify tandem arrays (genes ≤10 genes apart)
- Detect dispersed duplicates through all-by-all comparison
- Evolutionary Rate Analysis
- Calculate dN/dS for duplicate pairs
- Compare evolutionary rates between duplicated and singleton genes
- Assess selection patterns following duplication events
Functional Characterization Protocols
Expression Analysis
- Transcriptome Profiling
- Collect RNA-seq data across tissues, development stages, and stress conditions
- Quantify NBS-LRR expression levels (FPKM/TPM)
- Identify differentially expressed NBS-LRR genes under pathogen challenge
- Co-expression Network Analysis
- Perform weighted gene co-expression network analysis (WGCNA)
- Identify NBS-LRR genes hub nodes in immune networks
- Connect NBS-LRR genes to signaling pathways and metabolic processes [29]
Immune Function Validation
- Pathogen Response Assays
- Treat plants with defense hormones (e.g., salicylic acid)
- Monitor NBS-LRR gene induction through qRT-PCR and RNA-seq
- Identify key responsive genes through differential expression analysis [29]
- Genetic Transformation
- Overexpress candidate NBS-LRR genes in susceptible genotypes
- Knock out/down genes using CRISPR/Cas9 or RNAi
- Assess changes in pathogen resistance phenotypes
Table 3: Essential Research Reagents for Studying NBS-LRR Gene Evolution
| Reagent/Resource | Specific Examples | Application | Key Features |
|---|---|---|---|
| Genome Databases | NCBI Genome, Phytozome, Ensembl Plants | Genomic sequence retrieval | Chromosome-level assemblies, Annotation files |
| Domain Databases | Pfam, SMART, NCBI CDD, InterPro | Domain identification and verification | HMM profiles, Domain boundaries |
| Sequence Analysis Tools | HMMER v3, BLAST+ suite, MUSCLE, MAFFT | Sequence identification and alignment | Statistical rigor, Scalability |
| Phylogenetic Software | RAxML, IQ-TREE, MEGA, MrBayes | Evolutionary relationship inference | Maximum likelihood, Bayesian methods |
| Selection Analysis Programs | PAML (codeml), HyPhy, Selectome | dN/dS calculation, Selection detection | Branch-site models, False discovery control |
| Synteny Analysis Tools | MCScanX, SynVisio, D-GENIES | Microsynteny network construction | Visualization, Collinearity detection |
| Expression Databases | NCBI SRA, Expression Atlas, PlantRNA | Transcriptome data access | Multiple conditions, Differential expression |
| Plant Transformation Systems | Agrobacterium-mediated, Biolistics | Functional validation | Stable transformation, Transient expression |
The lineage-specific loss of TNL genes in monocots represents a compelling example of how evolutionary processes shape genomic architecture and functional capabilities in plant immune systems. The synthesis of evidence from multiple plant species reveals that this pattern resulted from an ancestral genomic deletion event followed by compensatory evolution through duplication and diversification of remaining NLR classes.
Future research directions should focus on:
- Elucidating compensatory mechanisms that maintain immune functionality in monocots despite TNL absence
- Exploring structural and functional convergence in CNL proteins that may compensate for lost TNL functions
- Investigating the impact of TNL loss on immune signaling networks and pathogen recognition capabilities
- Leveraging synteny-informed classification to resolve deeper evolutionary relationships in plant NLR genes
Understanding these lineage-specific evolutionary patterns provides fundamental insights into plant immunity evolution and offers potential strategies for engineering disease resistance in crop plants through manipulation of NLR gene repertoires.
From Sequence to Function: Methodologies for Identifying and Analyzing Duplicated NBS Genes
In plant genomes, the Nucleotide-Binding Site-Leucine-Rich Repeat (NBS-LRR) gene family represents one of the largest and most dynamic families of disease resistance genes. Research into their evolution is crucial for understanding plant immunity mechanisms and developing sustainable crop protection strategies. A fundamental driver of NBS-LRR diversity is gene duplication, which generates genetic novelty through mechanisms including tandem duplication, segmental duplication, and whole-genome duplication (WGD) [31] [23] [5]. These events create expanded gene families where subsequent evolutionary processes like neofunctionalization, subfunctionalization, or pseudogenization can occur [23] [5].
Studying these complex families requires precise identification and classification of their members. Bioinformatics pipelines that integrate Hidden Markov Models (HMMER), Pfam, and the Conserved Domain Database (CDD) have become the cornerstone for this work. These methods enable researchers to systematically identify, annotate, and classify genes across entire genomes, providing the foundational data for evolutionary analysis. This technical guide details the implementation of these core bioinformatic tools within the specific context of investigating gene duplication events in NBS gene evolution.
Core Tools and Databases for Domain Identification
The standard identification pipeline leverages three complementary tools to achieve a balance between sensitivity and specificity in detecting NBS domains and associated architectures.
- HMMER: Utilizes profile Hidden Markov Models (HMMs) for sensitive detection of remote homologs based on multiple sequence alignments. It is exceptionally powerful for identifying members of divergent protein families, such as the NBS-LRR family, by capturing conserved domain signatures even in sequences with low pairwise similarity [31] [32] [7].
- Pfam: A large collection of protein families, each represented by multiple sequence alignments and HMMs. The NB-ARC domain (PF00931) is the definitive model for the nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4, and serves as the primary query for identifying NBS-LRR genes [31] [5] [7].
- Conserved Domain Database (CDD): Curates domain models from multiple sources, including Pfam, and adds detailed annotation. CDD searches are crucial for validating domain presence, determining domain boundaries, and identifying associated N-terminal (TIR, CC, RPW8) and C-terminal (LRR) domains that define NBS-LRR subfamilies [31] [7].
The table below summarizes the role of each tool in a typical identification workflow.
Table 1: Core Bioinformatics Tools for NBS Gene Identification
| Tool | Primary Function | Key Input/Query | Typical Output | Role in NBS Gene Analysis |
|---|---|---|---|---|
| HMMER | Sequence homology search using profile HMMs | HMM profile (e.g., PF00931) & protein sequence file | List of significant domain hits with E-values | Initial, sensitive scan for NB-ARC domains in a proteome. |
| Pfam | Repository of protein family HMMs | HMM profile (e.g., PF00931) | Domain architecture & family classification | Provides the canonical model for the core NBS domain. |
| CDD | Domain annotation & validation | Protein sequence | Validated domain hits, boundaries, and classification | Confirms NBS domain and identifies flanking domains (TIR, CC, LRR). |
A successful genome-wide identification project relies on a suite of data and software resources. The table below lists key "research reagents" and their functions.
Table 2: Essential Research Reagents and Resources for NBS Gene Identification
| Resource Name | Type | Function in the Pipeline |
|---|---|---|
| NB-ARC (PF00931) | HMM Profile | Primary query for identifying the core NBS domain [31] [5]. |
| TIR (PF01582), CC, LRR profiles | HMM Profiles | Identification of N- and C-terminal domains for subfamily classification [5] [7]. |
| Reference Proteome | Data | The complete set of protein sequences for the organism of interest (e.g., from NCBI, Phytozome). |
| HMMER (v3.1b2+) | Software Suite | Executes the HMM search against the proteome using hmmscan [31] [7]. |
| NCBI's CDD | Web Service/Database | Validates HMM hits and refines domain boundaries via RPS-BLAST [31] [7]. |
| PlantCARE | Database | Used for subsequent promoter analysis (e.g., cis-regulatory element prediction) [31]. |
| MCScanX | Software | Identifies gene duplication types (tandem, segmental, WGD) from synteny data [31] [7]. |
Integrated Workflow for NBS Gene Identification and Classification
The following diagram illustrates the integrated bioinformatics pipeline, from data preparation to evolutionary analysis, highlighting how HMMER, Pfam, and CDD are combined.
Diagram 1: Integrated NBS Gene Identification and Analysis Workflow
Detailed Methodological Steps
The workflow can be broken down into the following detailed, sequential steps, as applied in recent studies:
- Data Retrieval: Obtain the complete proteome (all protein sequences) of the target species from a public database such as NCBI, Phytozome, or a dedicated genome database (e.g., Genome Database for Rosaceae) [5] [7].
- HMMER Scan with Pfam Model: Use the
hmmscancommand from the HMMER suite to scan the proteome against the NB-ARC HMM profile (PF00931). Studies typically use a relaxed E-value cutoff (e.g., 1.0) for the initial search to maximize sensitivity, capturing even divergent family members [31] [5]. - Candidate Generation: Combine results from the HMMER search and remove redundant entries to generate a non-redundant set of candidate NBS genes.
- CDD Validation and Domain Annotation: Submit the candidate protein sequences to NCBI's CDD search. This step is critical for confirming the presence and completeness of the NB-ARC domain (cd00204) and for identifying the presence of N-terminal (TIR, CC, RPW8) and C-terminal (LRR) domains. This annotation allows for the classification of genes into subfamilies (TNL, CNL, RNL, etc.) [31] [7].
- Final Curation: Manually inspect and curate the final list, removing any sequences where the NBS domain is truncated or otherwise incomplete.
Application in Gene Duplication Research: Experimental Protocols
The power of this bioinformatic pipeline is demonstrated by its application in identifying and characterizing gene duplication events. The following protocol, derived from a 2025 study on Capsicum annuum (pepper), exemplifies this approach [31].
Protocol: Identifying Tandem Duplications in the Pepper NLR Family
- Objective: To identify tandem duplication events contributing to the expansion of the NLR family in pepper and analyze their evolutionary and functional significance.
Bioinformatic Input: The high-quality 'Zhangshugang' reference genome of pepper and its annotation [31].
Step-by-Step Workflow:
- NLR Identification: The research group identified 288 high-confidence canonical NLR genes using the integrated HMMER/Pfam/CDD pipeline described above [31].
- Chromosomal Mapping: The physical positions of the identified NLR genes were mapped onto the chromosomes. Visualization revealed significant clustering, with a particularly high density in the telomeric regions of chromosomes 08 and 09 [31].
- Tandem Duplication Detection: Tandemly duplicated genes were defined as adjacent paralogs located within a specified distance on the same chromosome (e.g., separated by ≤1 intervening gene). Software tools like MCScanX are commonly used for this analysis [31] [7].
- Evolutionary Analysis: The contribution of tandem duplication to family expansion was quantified. In pepper, 53 of the 288 NLR genes (18.4%) were identified as tandem duplicates, predominantly on Chr08 and Chr09, establishing tandem duplication as the primary driver of NLR family expansion in this species [31].
- Functional Correlation: The expression of tandemly duplicated NLRs was investigated using RNA-seq data from Phophthora capsici-infected resistant and susceptible cultivars. This identified 44 significantly differentially expressed NLR genes, providing evidence for the functional role of these expanded clusters in pathogen response [31].
Key Findings and Output:
- Quantitative Result: Tandem duplication accounted for 18.4% of the pepper NLR genes.
- Genomic Distribution: Tandem clusters were enriched on specific chromosomes (Chr08, Chr09), often near telomeres.
- Functional Insight: Differentially expressed NLRs, including some from tandem arrays, were identified, with protein-protein interaction analysis suggesting certain members (e.g., Caz01g22900, Caz09g03820) may act as hubs in the immune network [31].
This protocol demonstrates how the initial gene identification pipeline feeds directly into sophisticated evolutionary genomics, directly addressing the role of gene duplication.
Protocol: Tracing Duplication in Allopolyploid Nicotiana tabacum
A 2025 study on Nicotiana (tobacco) provides another protocol for investigating the impact of whole-genome duplication (WGD) [7].
- Objective: To characterize the NBS gene families in allotetraploid N. tabacum and its diploid progenitors (N. sylvestris and N. tomentosiformis), tracing the origin of genes and the impact of WGD.
- Methods:
- The standard HMMER/CDD pipeline was applied to the three genomes, identifying 603 NBS genes in N. tabacum and 623 in its progenitors combined [7].
- Synteny Analysis: MCScanX was used to identify syntenic blocks between the N. tabacum genome and its parental genomes.
- Gene Tracing: By analyzing synteny, 76.62% of the NBS genes in N. tabacum could be traced back to their parental genome origin, demonstrating the profound impact of allopolyploidization on its NBS repertoire [7].
- Selection Pressure Analysis: The Ka/Ks ratio (non-synonymous to synonymous substitution rate) for duplicated gene pairs was calculated to infer the selective pressures acting on them post-duplication.
Data Interpretation and Integration with Evolutionary Analysis
The final stage of the pipeline involves interpreting the generated data to draw biological conclusions about NBS gene evolution.
- Quantifying Duplication Mechanisms: Research across species consistently shows that different duplication mechanisms contribute variably to NBS family expansion. The table below synthesizes findings from recent studies.
Table 3: Contribution of Different Duplication Mechanisms to NBS Family Expansion
| Species/Family | Tandem Duplication | Segmental/WGD | Evolutionary Pattern | Citation |
|---|---|---|---|---|
| Pepper (Capsicum annuum) | Primary driver (18.4% of genes) | Not specified | "Shrinking" pattern | [31] |
| Tobacco (Nicotiana tabacum) | Not primary | Major role (allotetraploidy) | "Expansion" via hybridization | [7] |
| Rosaceae Species (e.g., Apple, Peach) | Varies by species | Varies by species | Diverse patterns ("expansion & contraction") | [5] |
| Norway Spruce (Picea abies) | Widespread | Not specified | Involved in local adaptation | [23] |
- Linking Duplication to Function: A key advantage of this pipeline is its ability to connect evolutionary events to potential gene function. For example:
- Promoter Analysis: Analysis of promoter regions (e.g., using PlantCARE) in the pepper study revealed that 82.6% of NLR promoters contained binding sites for salicylic acid (SA) and/or jasmonic acid (JA) signaling, linking duplication to defense regulation [31].
- Expression Profiling: RNA-seq data from stress treatments can be mapped onto the identified NBS genes. Studies in cotton and Nicotiana have successfully identified specific NBS genes and orthogroups that are upregulated in response to pathogens, providing candidates for functional validation [20] [7].
The integrated use of HMMER, Pfam, and CDD forms a robust and essential bioinformatic pipeline for the accurate identification and classification of NBS-LRR genes. When this foundational data is fed into downstream analyses of synteny, phylogeny, and expression, it provides unparalleled insights into the evolutionary history of this critical gene family. By precisely quantifying the contributions of tandem, segmental, and whole-genome duplication events, researchers can unravel the complex "arms race" between plants and their pathogens, identifying key genetic elements that can be leveraged for future crop improvement.
In evolutionary genomics, the Ka/Ks ratio is a fundamental metric for quantifying the type of selection pressure acting on protein-coding genes. This ratio compares the rate of non-synonymous substitutions (Ka; changes the amino acid) to the rate of synonymous substitutions (Ks; does not change the amino acid). Synonymous substitutions often evolve neutrally, providing a baseline evolutionary rate. When Ka/Ks > 1, it indicates positive selection, where beneficial amino acid changes are driven by adaptive evolution. A Ka/Ks ≈ 1 signifies neutral evolution, while Ka/Ks < 1 suggests purifying selection, which removes deleterious mutations to conserve protein function [17] [19].
The analysis of Ka/Ks is particularly powerful when applied to duplicated genes, as it reveals the evolutionary forces shaping their fate post-duplication. Gene duplicates can undergo neofunctionalization (acquiring a new function), subfunctionalization (partitioning ancestral functions), or non-functionalization (becoming a pseudogene). Within the context of Nucleotide-Binding Site-Leucine-Rich Repeat (NBS-LRR) gene families—a cornerstone of the plant immune system—Ka/Ks analysis has been instrumental in deciphering the balance between evolutionary innovation and functional constraint [11] [17] [3]. For instance, studies on the maize ZmNBS gene family revealed that different duplication mechanisms are associated with distinct selection pressures: genes derived from whole-genome duplication (WGD) often exhibit strong purifying selection (low Ka/Ks), whereas those from tandem and proximal duplications frequently show signs of relaxed or positive selection, highlighting their role in adaptive evolution [11].
Theoretical Framework: Ka/Ks and Selection Pressures
Interpretation of Ka/Ks Values
The table below summarizes the standard interpretation of Ka/Ks ratios.
| Ka/Ks Value | Type of Selection | Evolutionary Interpretation |
|---|---|---|
| > 1 | Positive/Diversifying Selection | Amino acid changes are advantageous, driving adaptive evolution. Common in genes involved in arms races (e.g., plant-pathogen interactions) [11]. |
| ≈ 1 | Neutral Evolution | Mutations are fixed without selective constraint; genes evolve at the expected rate. |
| < 1 | Purifying/Stabilizing Selection | Deleterious amino acid changes are removed; the gene is under functional constraint [11] [17] [19]. |
Ka/Ks Application in NBS Gene Evolution
Research on NBS-LRR genes across diverse species, including maize, Nicotiana, and Rosaceae, consistently shows that these disease-resistance genes are often governed by a "birth-and-death" evolutionary model [11] [3]. Different modes of gene duplication are subject to varying selective pressures, which can be quantified by Ka/Ks:
- Tandem Duplications: Often associated with adaptive evolution, showing higher Ka/Ks values. This is critical for generating diversity in pathogen recognition [11] [17].
- Whole-Genome Duplications (WGD/Polyploidy): Typically under strong purifying selection (low Ka/Ks), preserving core immune functions [11] [33] [17]. Analysis in Nicotiana species found that WGD significantly contributed to NBS family expansion, with most duplicates under purifying selection [17].
Computational Methodology: A Step-by-Step Guide
This section provides a detailed protocol for calculating Ka/Ks ratios for duplicated gene pairs, incorporating tools and practices from recent genomic studies.
The following diagram illustrates the end-to-end computational workflow for Ka/Ks analysis.
Detailed Experimental Protocols
Step 1: Identify Duplicated Gene Pairs
- Method: Use tools like MCScanX to perform intra-genomic self-BLASTP and identify segmental and tandem duplications across the whole genome [17] [19].
- Rationale: This provides the evolutionary context (duplication mode) essential for interpreting Ka/Ks results.
Step 2: Sequence Retrieval and Preparation
- Input Data: Obtain the Coding DNA Sequences (CDS) and their corresponding protein sequences for the identified gene pairs from genomic annotation files (GFF/GTF format).
- Tool:
TBtoolscan be used for efficient batch extraction from GFF3 files [17] [19].
Step 3: Multiple Sequence Alignment
- Protein Alignment: First, align the protein sequences using MUSCLE or MAFFT. Protein alignment is more accurate than nucleotide alignment over evolutionary distances.
- CDS Alignment: Use the protein alignment as a guide to align the corresponding CDS sequences, ensuring codons remain intact. The
ParaATtool can automate this process [17].
Step 4: Ka/Ks Calculation
- Tool: KaKs_Calculator 2.0 is a standard tool that implements multiple models for calculation (e.g., Nei-Gojobori (NG), Yang-Nielsen (YN)) [17] [19].
- Command Example:
- Model Selection: The Nei-Gojobori (NG) model is commonly used for its simplicity and robustness [17].
Key Research Reagents and Computational Tools
The table below catalogs essential reagents and software tools for conducting Ka/Ks analysis.
| Item Name | Type/Category | Function in Ka/Ks Analysis |
|---|---|---|
| MCScanX [17] [19] | Software | Identifies collinear genomic blocks and classifies gene duplication modes (WGD, segmental, tandem). |
| MUSCLE [17] | Software | Performs high-accuracy multiple sequence alignment of protein sequences. |
| ParaAT [17] | Software | Automates the alignment of CDS sequences based on their corresponding protein sequence alignment. |
| KaKs_Calculator 2.0 [17] [19] | Software | Calculates Ka, Ks, and Ka/Ks values from aligned CDS sequences using various evolutionary models. |
| CDS & Protein Sequences | Data | The primary input data, retrieved from genome annotation files. |
| Genome Annotation File (GFF/GTF) | Data | Provides the structural information (exon coordinates, reading frame) needed to extract correct CDS. |
Data Analysis and Interpretation in NBS Gene Research
Summarizing and Presenting Ka/Ks Data
After calculation, results should be compiled for comparative analysis. The following table exemplifies how Ka/Ks data can be structured for different duplication types, using patterns observed in NBS gene studies.
| Duplication Mechanism | Typical Ka/Ks Range | Inferred Selection Pressure | Biological Implication in NBS Genes |
|---|---|---|---|
| Whole-Genome Duplication (WGD) | < 1 (Low) [11] | Strong Purifying Selection | Conserves core immune functions; stable "core" NBS subgroups [11] [17]. |
| Tandem Duplication (TD) | Often closer to 1 or >1 [11] | Relaxed or Positive Selection | Drives diversification for new pathogen recognition; "adaptive" NBS subgroups [11] [3]. |
| Segmental Duplication | Variable | Purifying to Relaxed | Can contribute to both conservation and diversification of gene families. |
Case Studies and Research Context
- Maize ZmNBS Genes: A pan-genomic analysis revealed a "core-adaptive" model. Conserved "core" genes (e.g., ZmNBS31) showed evidence of purifying selection, while variable "adaptive" subgroups (e.g., ZmNBS1-10) experienced relaxed or positive selection, linked to tandem duplications [11].
- Nicotiana NBS Genes: Analysis of three tobacco genomes found that whole-genome duplication was a major expansion force, with most gene duplicates under purifying selection (Ka/Ks < 1) [17].
- Rosaceae NBS-LRR Genes: Studies across 12 species demonstrated that independent gene duplication and loss events led to distinct evolutionary patterns (e.g., "expansion and contraction"), which Ka/Ks analysis helps to decipher [3].
Advanced Analysis: Decision Framework and Visualization
Interpreting Ka/Ks results requires considering statistical confidence and biological context. The following decision diagram outlines this process.
Key Considerations for Robust Analysis
- Statistical Significance: Use statistical tests (e.g., Fisher's exact test) available in KaKs_Calculator to determine if Ka/Ks > 1 is significant.
- Ks Saturation: For very ancient duplicates, Ks values can saturate, making Ka/Ks unreliable. Filter out pairs with Ks > 2-3 for more accurate results.
- Biological Context: Always integrate Ka/Ks findings with other evidence, such as gene expression data under stress [11] [17], protein structure, and phylogenetic analysis. A gene under purifying selection overall may have a few sites under positive selection, detectable by site-specific models (e.g., PAML).
Gene duplication is a fundamental evolutionary process that provides the raw genetic material for functional innovation and adaptation in organisms [34]. In plant genomes, genes involved in pathogen resistance, such as the Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) genes, are frequently observed to undergo extensive duplication events [14] [5]. These genes constitute one of the largest gene families in plants and play a critical role in detecting pathogen effectors and initiating immune responses [5] [35]. The evolution of these gene families is characterized by dynamic patterns of expansion and contraction, driven by various duplication mechanisms including tandem duplication, segmental duplication, and whole-genome duplication (WGD) [5] [34].
Understanding these duplication events requires specialized bioinformatic tools that can detect and analyze syntenic and collinear regions across genomes. MCScanX is a comprehensive toolkit specifically designed for this purpose, implementing an adjusted MCScan algorithm for detecting synteny and collinearity with enhanced analytical capabilities [36] [37] [38]. This technical guide provides an in-depth overview of utilizing MCScanX for duplication event detection, with particular emphasis on its application in evolutionary studies of NBS-LRR genes and other duplication-prone gene families involved in evolutionary arms races.
Core Components and Installation
MCScanX consists of two primary components: a modified version of the MCScan algorithm optimized for user convenience and visualization of syntenic blocks, and a suite of downstream analysis tools for diverse biological investigations [36]. The software is designed for command-line execution on Linux and Mac OS systems, with all programs including built-in usage information accessible by running them without parameters [36].
Installation follows a standard compilation process:
The package generates multiple executable programs, including the main MCScanX application, MCScanXh for alternative homology input formats, and duplicategene_classifier for determining duplication origins [36]. Additionally, twelve downstream analysis tools provide specialized functionalities for tandem array detection, visualization, and evolutionary analysis [36].
Input File Preparation
Proper input file preparation is crucial for successful MCScanX analysis. The tool requires two primary input files:
BLASTP Output File (.blast): An all-against-all BLASTP search result in tabular format (m8), typically generated with parameters:
-e 1e-10 -b 5 -v 5 -m 8[36]. For optimal results, the number of BLASTP hits per gene should be restricted to approximately the top 5 matches [36].Gene Position File (.gff or .bed): A tab-delimited file containing gene positions following the format:
chr# start_position end_position gene[36]. Chromosome identifiers should use a two-letter species prefix (e.g., "at2" for Arabidopsis thaliana chromosome 2). The file must not contain duplicate gene entries [36].
For multi-genome comparisons, both intra-species and inter-species BLAST results and gene positions are concatenated into single input files [36].
Table 1: Key Research Reagent Solutions for MCScanX Analysis
| Research Reagent | Function | Source/Implementation |
|---|---|---|
| BLAST+ Suite | Generate protein sequence similarity data for synteny detection | NCBI [39] [36] |
| MCScanX | Core synteny and collinearity detection algorithm | GitHub Repository [36] |
| InterProScan | Protein domain annotation for functional validation | EBI [39] [40] |
| KEGG Tools | Pathway-based categorization of duplicated genes | KEGG Database [39] [40] |
| Duplicategeneclassifier | Categorize gene duplication modes (WGD, tandem, proximal, dispersed) | MCScanX Package [36] |
Experimental Protocol for NBS Gene Duplication Analysis
Workflow for Synteny Detection
The following diagram illustrates the complete workflow for detecting duplication events in NBS genes using MCScanX:
Executing MCScanX
With properly prepared input files, execute MCScanX using the command:
where directory/prefix specifies the location and prefix for input files (e.g., files named prefix.blast and prefix.gff) [36].
MCScanX provides several advanced parameters for tuning synteny detection:
-k: Match score (default: 50)-g: Gap penalty (default: -1)-s: Minimum genes required to call synteny (default: 5)-e: E-value threshold (default: 1e-5)-m: Maximum gaps allowed (default: 20) [36]
For studying NBS gene evolution, adjusting the -s parameter to lower values (3-5 genes) may help detect smaller syntenic blocks characteristic of rapidly evolving resistance gene clusters [14] [5].
Downstream Analysis of Duplication Events
MCScanX includes specialized tools for detailed analysis of duplication events:
Duplicate Gene Classification:
This program categorizes genes into five classes: singleton (0), dispersed (1), proximal (2), tandem (3), and segmental/WGD (4) [36]. This classification is particularly valuable for understanding the predominant mechanisms driving NBS-LRR gene family expansion in specific lineages [5].
Syntenic Tandem Array Detection:
This tool identifies tandem duplications within syntenic blocks, which is relevant for studying NBS-LRR genes as they frequently form tandem arrays [14] [36].
Data Interpretation in NBS Gene Evolution Context
Analyzing MCScanX Output
MCScanX generates two primary output types:
Synteny Text File (.synteny): Contains pairwise synteny blocks with alignment scores, e-values, and gene-by-gene correspondences [36].
HTML Visualization Directory: Provides chromosome-based visualizations of syntenic blocks, with tandem genes highlighted in red [36].
For NBS gene family analysis, researchers should particularly note:
- Clustered arrangements of NBS genes, as over 22% of NBS genes occur in clusters in some species [35].
- Differential evolutionary patterns across lineages, such as the "first expansion and then contraction" pattern observed in Rubus occidentalis versus "continuous expansion" in Rosa chinensis [5].
- Association with duplication-inducing elements, as arms-race genes like NBS-LRRs often associate with duplication-prone genomic regions [14].
Evolutionary Analysis of NBS Gene Duplication
The following diagram illustrates the evolutionary interpretation framework for NBS gene duplication analysis:
Table 2: MCScanX Parameters for Optimizing NBS Gene Duplication Detection
| Parameter | Default Value | Recommended for NBS Genes | Rationale |
|---|---|---|---|
| MATCH_SIZE (-s) | 5 genes | 3-5 genes | NBS clusters may form smaller syntenic blocks |
| E_VALUE (-e) | 1e-5 | 1e-5 to 1e-10 | Balance sensitivity and specificity |
| GAP_PENALTY (-g) | -1 | -0.5 to -2 | Accommodate higher rearrangement rates in R-genes |
| MAX_GAPS (-m) | 20 | 25-30 | Account for sequence divergence in arms-race genes |
Case Study: Rosaceae NBS-LRR Gene Evolution
Application of MCScanX in Comparative Genomics
A comprehensive study of NBS-LRR genes across 12 Rosaceae species utilized synteny-based approaches to reveal distinct evolutionary patterns [5]. The research identified 2,188 NBS-LRR genes with remarkable variation in gene numbers across species, attributable to independent gene duplication and loss events [5].
The application of MCScanX in this context enabled researchers to:
- Reconstruct ancestral genes, identifying 102 ancestral NBS-LRR genes (7 RNLs, 26 TNLs, and 69 CNLs) [5].
- Trace lineage-specific trajectories, revealing patterns such as "first expansion and then contraction" in Rubus occidentalis and Potentilla micrantha versus "continuous expansion" in Rosa chinensis [5].
- Correlate duplication mechanisms with evolutionary outcomes, showing how different Rosaceae lineages employed varying duplication strategies (tandem, segmental, WGD) to adapt to pathogen pressures [5].
Technical Considerations for NBS Gene Studies
When applying MCScanX specifically to NBS gene evolution, several technical considerations enhance results:
Pre-processing for NBS Identification:
- Combine BLAST and HMMER searches using the NB-ARC domain (PF00931) to identify candidate NBS-LRR genes [5] [35].
- Validate N-terminal domains (TIR, CC, RPW8) using Pfam and NCBI-CDD with an E-value cutoff of 10⁻⁴ [5].
- Classify genes into TNL, CNL, and RNL subclasses before synteny analysis [5].
Evolutionary Rate Considerations:
- NBS-LRR genes typically exhibit higher substitution rates and more frequent recombination between paralogs [5].
- Adjust alignment parameters to accommodate elevated evolutionary rates while maintaining detection sensitivity.
Integration with Complementary Tools
Expanding Analytical Capabilities
While MCScanX provides comprehensive synteny detection, integration with specialized tools enhances duplication analysis:
HSDFinder Integration: For identifying highly similar duplicated genes (HSDs) with ≥90% pairwise identity, HSDFinder offers complementary functionality [39] [40]. The tool categorizes duplicates using Pfam domains and KEGG pathways, generating heatmap visualizations across species [39] [40]. This approach is particularly valuable for detecting recent duplication events in NBS genes that may contribute to gene dosage effects in stress adaptation [40].
Visualization Enhancements:
- Circos plots: Generate circular representations of syntenic relationships [36] [38].
- Dual synteny plots: Visualize relationships between three genomes simultaneously [36].
- Dot plots: Display gene correspondences between chromosomal regions [36].
Validation and Functional Analysis
Confirm MCScanX findings through:
- Phylogenetic analysis of NBS gene families to validate duplication events [5].
- Motif analysis using MEME suite to identify conserved domain architectures [5] [35].
- Expression correlation for tandemly duplicated NBS genes to identify potential dosage effects [14].
MCScanX provides an powerful computational framework for detecting and analyzing gene duplication events through synteny and collinearity analysis. Its application to NBS gene evolution research has revealed the dynamic and complex evolutionary patterns underlying plant pathogen resistance mechanisms. The toolkit's ability to classify duplication modes, visualize syntenic relationships, and facilitate comparative genomics makes it an indispensable resource for researchers investigating gene family evolution, particularly in the context of arms-race genes subject to rapid diversification through duplication events. As genomic data continues to expand, MCScanX remains a critical tool for deciphering the duplication histories that shape genome evolution and functional adaptation.
This technical guide explores the integration of RNA sequencing (RNA-seq) methodologies to investigate how gene duplication events influence transcriptional regulation, with a specific focus on the evolution of Nucleotide-Binding Site Leucine-Rich Repeat (NBS-LRR) genes. Gene duplication serves as a primary source of evolutionary innovation, creating genetic material that can diverge in function and regulation. The advent of high-throughput transcriptomics has enabled researchers to decode the complex regulatory consequences of duplication events, providing insights into gene family expansion, functional diversification, and the molecular basis of adaptive traits. This whitepaper details experimental and computational frameworks for analyzing duplicate gene expression, outlining protocols for transcriptome assembly, differential expression analysis, and co-expression network construction specifically tailored for polyploid genomes and complex gene families.
Gene duplication is a fundamental evolutionary process that generates genetic novelty, with duplicated genes serving as primary sources for functional innovation and specialized adaptation. In plant genomes, whole-genome duplication (WGD) events are particularly prevalent and have driven the expansion of many multigene families, including disease-resistant NBS-LRR genes [41]. Following duplication, genes can undergo several evolutionary trajectories: nonfunctionalization (loss of function), neofunctionalization (acquisition of new function), or subfunctionalization (partitioning of ancestral functions) [42].
The development of RNA-seq technologies has revolutionized our ability to study the transcriptional consequences of gene duplication. By providing a quantitative snapshot of the transcriptome, researchers can now investigate how duplication events lead to expression partitioning, homoeolog bias, and regulatory divergence [41]. For NBS-LRR genes, which play crucial roles in plant immunity through effector-triggered immunity, understanding these regulatory dynamics has significant implications for crop improvement and disease resistance breeding [43].
RNA-Seq Experimental Design for Duplication Studies
Special Considerations for Polyploid Genomes and Gene Families
Studies investigating duplicated genes, particularly in polyploid organisms, require specific experimental considerations:
- Sequence Ambiguity: The increased ambiguity introduced by duplicated sequences complicates read alignment and transcript quantification. Specialized tools that account for multiple homoeologs are essential [41].
- Subgenome Resolution: In allopolyploids, maintenance of subgenome-specific references enables investigation of homoeolog expression bias - unequal expression contributions from duplicated genes [41].
- Tissue-Specific Sampling: As homoeolog silencing effects can be non-uniform across tissues, strategic sampling of multiple tissues and developmental stages is critical for comprehensive expression profiling [41].
Minimizing Technical Variation
Appropriate experimental design is crucial for generating meaningful RNA-seq data. Key strategies to minimize batch effects include:
Table 1: Strategies to Mitigate Batch Effects in RNA-Seq Experiments
| Source of Variation | Mitigation Strategy |
|---|---|
| Temporal Effects | Process all samples simultaneously; harvest at same time of day |
| Technical Handling | Use a single researcher for procedures; minimize freeze-thaw cycles |
| Sequencing Effects | Sequence all samples in a single run; balance experimental groups across lanes |
| Biological Variation | Use littermate or intra-animal controls; increase biological replicates |
Batch effects can obscure true biological signals and must be controlled throughout the experimental process [44]. Biological replicates (typically n≥3) are essential for robust statistical analysis of differential expression.
Computational Analysis of Duplicated Gene Expression
RNA-Seq Data Processing Workflow
The initial phase of RNA-seq analysis involves transforming raw sequencing data into quantitative gene expression measurements. The standard workflow consists of:
- Read Trimming and Quality Control: Remove adapter contamination and low-quality bases using tools like Trimmomatic, then verify sequence quality with FastQC [45].
- Alignment to Reference Genome: Map reads to a reference genome using splice-aware aligners such as HiSat2 or STAR, which is particularly important for eukaryotic transcripts [45] [46].
- Read Quantification: Generate raw count data using tools like HTSeq, assigning reads to genomic features while accounting for overlapping genes [45] [44].
For studies of duplicated genes, special consideration must be given to the alignment and quantification steps, as standard methods may misassign reads from highly similar paralogs. Approaches include using subgenome-specific references for polyploids or pseudogenome references that represent all known haplotypes [41].
Identification of Duplicated Genes
Before expression analysis, a comprehensive catalog of duplicated genes must be established. For NBS-LRR genes, this involves:
- Hidden Markov Model (HMM) Searches: Use the NB-ARC domain (PF00931) as query to identify NBS-containing genes in the genome [43].
- Domain Architecture Analysis: Annotate protein domains (CC, TIR, LRR) using tools like Pfam and CDD to classify NBS-LRR genes into subfamilies (CNL, TNL, RNL) [47] [43].
- Synteny and Phylogenetic Analysis: Identify orthologous and paralogous relationships through comparative genomics and maximum likelihood phylogenetics [43].
In barley (Hordeum vulgare), 96 NBS-encoding genes were identified through such methods, with 53.1% classified as NBS-LRR, 14.6% as CC-NBS-LRR, 26% as NBS, and 6.3% as CC-NBS [47]. In rye (Secale cereale), 582 NBS-LRR genes were identified, with chromosome 4 containing the largest number, suggesting chromosome-specific expansion patterns [43].
Differential Expression Analysis
Differential expression analysis identifies genes with statistically significant expression changes between conditions. The standard approach involves:
- Count Normalization: Account for library size and composition biases using methods like TMM (Trimmed Mean of M-values) in edgeR or median-of-ratios in DESeq2 [44].
- Statistical Testing: Employ negative binomial models in tools like DESeq2 or edgeR to identify differentially expressed genes (DEGs) [45] [46].
- Multiple Testing Correction: Apply Benjamini-Hochberg false discovery rate (FDR) control to account for the thousands of simultaneous statistical tests [45].
For studies of duplicated genes, additional considerations include testing for expression-level dominance (where the combined expression of duplicates matches one parent) and transgressive expression (where expression exceeds both parents) [41].
Table 2: Key Differential Expression Analysis Tools
| Tool | Statistical Approach | Strengths | Considerations for Duplicated Genes |
|---|---|---|---|
| DESeq2 | Negative binomial generalized linear model | Robust with small sample sizes; conservative | Requires careful model specification for complex designs |
| edgeR | Negative binomial models with empirical Bayes | Good performance with replicates | Similar considerations as DESeq2 |
| limma-voom | Linear modeling with precision weights | Fast; good for large experiments | Assumes normality after transformation |
Co-expression Network Analysis
Co-expression network analysis identifies sets of genes with correlated expression patterns across samples, potentially revealing functional relationships and coordinated regulation. The standard approach involves:
- Correlation Calculation: Compute pairwise correlation coefficients (e.g., Pearson or Spearman) between gene expression profiles [45] [46].
- Network Construction: Identify significant co-expression relationships using thresholds (e.g., Pearson correlation > 0.99, FDR < 0.01) [45].
- Module Detection: Identify densely connected groups of genes (modules) using clustering algorithms [48].
In a study of nanocurcumin-treated colorectal cancer cells, researchers identified 14,472 significant co-expression relationships between 20 key lncRNAs and 70,711 mRNAs, revealing extensive regulatory networks [45]. Similarly, in fire ant queens, DEL:DEG pairs with high association (Spearman's |rho| > 0.8, p-value < 0.01) revealed coordinated regulation during reproductive transition [46].
Case Study: NBS-LRR Gene Family Evolution and Expression
Genomic Distribution and Tandem Duplication
NBS-LRR genes frequently occur in genomic clusters resulting from tandem duplication events. In barley, 85 NBS-encoding genes were mapped across the seven chromosomes, with 50% located on chromosomes 7H, 2H, and 3H, showing a tendency to cluster in distal telomeric regions [47]. Nine gene clusters representing 22.35% of mapped barley NBS-encoding genes were identified, demonstrating that tandem duplication represents an important mechanism for the expansion of this gene family [47].
Comparative genomics reveals species-specific expansion patterns. The rye genome contains 582 NBS-LRR genes, exceeding the numbers found in barley and diploid wheat genomes [43]. Phylogenetic analysis suggests that at least 740 NBS-LRR lineages were present in the common ancestor of rye, barley, and Triticum urartu, but most have been inherited by only one or two species, with just 65 preserved in all three species [43]. This pattern highlights the dynamic birth-and-death evolution of NBS-LRR genes, with frequent duplication and loss events shaping the repertoire in each lineage.
Expression Divergence in Duplicated NBS-LRR Genes
Following duplication, NBS-LRR genes can diverge in their expression patterns. In barley, 87 out of 96 identified NBS genes showed expression evidence, exhibiting various and quantitatively uneven expression patterns across distinct tissues, organs, and development stages [47]. This expression heterogeneity suggests subfunctionalization or neofunctionalization of duplicated NBS-LRR genes.
Regulatory divergence can also occur through alternative splicing. In Arabidopsis, approximately 30% of alternative splicing events in α-whole-genome duplicates and 33% in tandem duplicates are qualitatively conserved within leaf tissue [42]. However, only 31% of shared AS events in α-whole-genome duplicates and 41% in tandem duplicates had similar frequencies in both paralogs, indicating considerable quantitative divergence in post-transcriptional regulation [42].
Visualization and Interpretation of Expression Data
Heatmaps and Clustering Visualization
Heatmaps provide an intuitive visualization of gene expression patterns across samples and are particularly useful for identifying co-expressed gene clusters. Effective heatmap generation involves:
- Data Scaling: Apply Z-score normalization to make expression patterns comparable across genes with different expression levels [49].
- Distance Metrics: Select appropriate distance measures (e.g., Euclidean, Manhattan, or correlation-based distances) for clustering [49].
- Clustering Algorithms: Choose hierarchical, k-means, or other clustering methods to group genes with similar expression profiles [49].
Tools like pheatmap in R provide comprehensive heatmap generation capabilities with built-in scaling functions and customization options for publication-quality figures [49]. For large datasets, such as those containing hundreds of duplicated genes, interactive heatmaps using heatmaply allow researchers to explore individual data points by mousing over tiles [49].
Functional Enrichment Analysis
Functional enrichment analysis places expression results in biological context by identifying over-represented functional categories. Standard approaches include:
- Gene Ontology (GO) Enrichment: Identify enriched biological processes, molecular functions, and cellular components among differentially expressed genes [45] [48].
- KEGG Pathway Analysis: Detect enriched metabolic and signaling pathways to understand the broader functional implications of expression changes [45].
- Statistical Assessment: Use hypergeometric tests or Fisher's exact test with multiple testing correction to identify statistically significant enrichment [45].
In studies of nanocurcumin-treated cancer cells, functional enrichment analysis revealed that modulated lncRNAs and their targets participate in cell cycle, p53 signaling, translation, and helicase activity pathways [45]. Similarly, in adult degenerative scoliosis, GO analysis indicated that lncRNA-targeted genes participate in AMPK signaling, lysosomal function, and ubiquitin-mediated proteolysis [48].
Table 3: Key Research Reagents and Computational Tools for RNA-Seq Analysis of Duplicated Genes
| Category | Specific Tools/Reagents | Application | Considerations |
|---|---|---|---|
| Library Preparation | TruSeq RNA Sample Prep Kit; NEBNext Ultra DNA Library Prep Kit | cDNA library construction for sequencing | Poly(A) selection for mRNA; rRNA depletion for total RNA |
| Sequencing Platforms | Illumina HiSeq 4000; NextSeq 500 | High-throughput sequencing | Read length (75-150bp), single vs paired-end, coverage depth |
| Alignment Tools | HiSat2; STAR; TopHat2 | Mapping reads to reference genome | Splice-awareness crucial for eukaryotes; specialized mappers for polyploids |
| Quantification Tools | HTSeq; featureCounts; Salmon | Generating expression counts | Resolution of multi-mapping reads critical for duplicated genes |
| Differential Expression | DESeq2; edgeR; limma-voom | Identifying statistically significant expression changes | Appropriate experimental design and replication essential |
| Specialized Polyploid Tools | PolyCat; HomeoRoq; SPIA | Handling multi-genome references | Subgenome-specific alignment and quantification |
RNA-seq technologies have fundamentally transformed our ability to link gene duplication events with transcriptional regulation, providing unprecedented insights into the evolutionary dynamics of gene families. For NBS-LRR genes and other duplicated gene families, integrated genomic and transcriptomic approaches have revealed complex patterns of expression divergence, regulatory innovation, and functional specialization.
Future advancements in this field will likely come from several technological developments:
- Long-read sequencing will improve resolution of complex genomic regions with tandem duplicates
- Single-cell RNA-seq will enable investigation of expression variation in duplicated genes at cellular resolution
- Integrated multi-omics approaches will connect transcriptional regulation with epigenetic modifications in duplicated genes
- Machine learning applications will help predict functional fates and regulatory outcomes from sequence features of duplicates
As these methodologies mature, researchers will gain increasingly sophisticated tools to decipher the complex relationship between gene duplication and transcriptional regulation, with significant implications for understanding evolutionary processes and engineering improved crop varieties with enhanced disease resistance.
Navigating Analytical Challenges: Refining NBS Gene Annotation and Functional Prediction
The accurate identification and annotation of genes form the critical foundation upon which virtually all downstream genomic analyses are built. However, researchers consistently face significant challenges stemming from genome incompleteness and the misannotation of pseudogenes as functional genes. In the context of studying gene duplication events, particularly in rapidly evolving families like nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes, these annotation errors can profoundly distort evolutionary interpretations and functional analyses. Draft genome assemblies frequently contain substantial errors in gene number estimates, with studies revealing that upwards of 40% of all gene families may be inferred to have the wrong number of genes in draft assemblies [50]. These inaccuracies arise from multiple sources, including genome fragmentation that splits single genes across multiple contigs, haplotype divergence in heterozygous individuals being misinterpreted as separate loci, and the collapsing of recent paralogs into single consensus sequences [50].
The problem is particularly acute when studying disease resistance (R) genes in plants, especially the NBS-LRR family, which evolves rapidly through duplication events and exhibits substantial copy number variation among species. For example, comparative analyses across six Fragaria species identified 1,134 NBS-LRR genes comprising 184 gene families, with lineage-specific duplications occurring before species divergence [9]. Without accurate annotation methods to distinguish functional genes from pseudogenes and to properly identify orthologs and paralogs, researchers risk drawing erroneous conclusions about evolutionary history, selective pressures, and functional diversity. This technical guide provides comprehensive strategies for addressing these challenges, with specific applications to NBS gene evolution research.
Genome Assembly Quality Issues
The quality of genome assembly fundamentally constrains annotation accuracy. Low-quality assemblies introduce numerous artifacts that directly impact gene copy number estimation and functional annotation. Draft assemblies are particularly problematic for gene-dense regions or complex gene families with high sequence similarity between members. Sequence fragmentation causes single genes to be "cleaved" across multiple contigs or scaffolds, leading to the artificial inflation of gene numbers as each fragment may be annotated as a separate gene [50]. Conversely, haplotype collapse occurs when heterozygous regions are assembled as a single consensus sequence, thereby obscuring true genetic variation and potentially missing functional genes [50] [51]. In repetitive regions, such as tandemly duplicated NBS-LRR clusters, these problems are exacerbated, with studies showing that more than 50% of genes may have the wrong number of copies in draft genomes [50].
The choice of sequencing technology and assembly algorithms significantly impacts these error rates. Long-read sequencing technologies have improved assembly contiguity, but challenges remain, particularly in highly duplicated regions. In polar fish genomes, for instance, repetitive antifreeze protein (AFP) gene arrays present substantial assembly challenges, with assembly uncertainty being "ubiquitous across AFP array haplotypes" [51]. Similarly, in plant genomes, NBS-LRR genes are often arranged in tandem clusters that are difficult to resolve accurately [52].
Pseudogene Misannotation Challenges
Pseudogenes present particularly difficult challenges for annotation pipelines. These genomic sequences resemble functional genes but contain disabling mutations that prevent production of functional proteins. Two primary categories exist: processed pseudogenes (reverse-transcribed from mRNA and reintegrated into the genome, lacking introns) and non-processed pseudogenes (originating from gene duplication events that subsequently accumulated disabling mutations) [53] [54]. The accurate identification of pseudogenes is crucial for correct gene counts and evolutionary analyses, yet standard annotation pipelines often misannotate them as functional genes.
Several factors contribute to pseudogene misannotation. Transcribed pseudogenes are especially problematic because their expression evidence can be misinterpreted as support for functionality [53]. Additionally, non-processed pseudogenes that retain aspects of exon-intron structure can be mistakenly incorporated into gene annotations. One study examining the Ensembl human gene predictions found that 9% of genes (2,011 genes) were likely pseudogenes based on expression evidence profiling, with approximately 40% of these displaying multi-exon structures characteristic of non-processed pseudogenes [53]. For NBS-LRR genes, which frequently undergo duplication and pseudogenization, this misclassification can significantly inflate functional gene counts and obscure evolutionary patterns.
Inconsistencies Across Annotation Pipelines
Different annotation methods and pipelines can yield markedly distinct gene models and repertoires for the same genome. A recent investigation into the effect of structural gene annotation on orthology inference revealed "significant discrepancies between sources" when comparing gene models from NCBI, Ensembl, UniProt, and Augustus [55]. These inconsistencies directly impact downstream comparative analyses, including orthology assignments and evolutionary interpretations.
The problem is particularly pronounced for non-model organisms where transcriptomic evidence may be limited, forcing greater reliance on ab initio prediction tools. Without community standards for annotation, most published gene annotations result from ad hoc pipelines, leading to heterogeneity that complicates cross-study comparisons [55]. For researchers studying gene family evolution, these inconsistencies can create artificial patterns of lineage-specific expansion or contraction, especially in rapidly evolving families like NBS-LRR genes where species-specific duplications are common [9].
Table 1: Major Sources of Annotation Errors and Their Impact on Gene Family Analysis
| Error Source | Impact on Gene Count | Effect on Evolutionary Analyses | Prevalence in NBS-LRR Genes |
|---|---|---|---|
| Genome Fragmentation | Artificial inflation (cleaved genes) | Overestimation of gene family size | High in clustered arrangements |
| Haplotype Collapse | Underestimation of diversity | Missing recent duplications | Moderate in heterozygous species |
| Pseudogene Misannotation | Overestimation of functional genes | Incorrect inference of functional evolution | Very high due to rapid turnover |
| Pipeline Inconsistencies | Variable counts between studies | Reduced comparability across studies | High across all analyses |
Detection and Mitigation Strategies
Computational Approaches for Pseudogene Identification
Robust computational methods are essential for distinguishing functional genes from pseudogenes. Effective pipelines typically combine similarity searches with disablement detection. A comprehensive approach for identifying pseudogenes should include:
Whole genome profiling of expression evidence: This method involves mapping existing transcript and protein sequences to the genome and identifying discrepancies that indicate pseudogenization. The process includes identifying "best hits" for every sequence aligned to the genome, requiring ≥98% identity with ≥90% coverage of the original sequence [53]. Sequences that align to multiple locations but show disablements (frameshifts, in-frame stop codons) at secondary locations indicate potential pseudogenes.
Structure-based classification: This approach explicitly uses intron-exon structure from putative parent genes to classify pseudogenes [54]. The method involves two complementary routines: one focusing on processed pseudogenes (using full-length proteins as queries) and another focusing on duplicated pseudogenes (using individual exons as queries). Alignments between duplicated pseudogenes and their parents must span intron-exon junctions to distinguish true duplicated pseudogenes from processed pseudogenes with insertions.
Integrated disablement detection: This combines BLAST searches with refined tools like GeneWise to detect frameshifts and in-frame stop codons in putative coding regions. The frameshift rate is calculated as the sum of frameshifts divided by the sum of match length, providing a quantitative measure of functionality [53].
For NBS-LRR genes specifically, additional criteria should be applied, including verification of conserved NBS domain motifs (P-loop, GLPL, Kinase-2, RNBS-B) and assessment of integrated domains that may indicate functional specialization [52].
Improving Genome Assembly and Annotation Quality
Enhancing the initial genome assembly is fundamental to reducing annotation errors. Several strategies have proven effective:
Hybrid sequencing approaches: Combining long-read technologies (PacBio HiFi, Oxford Nanopore) with chromatin conformation data (Hi-C) significantly improves assembly contiguity and phasing accuracy. For polar fish genomes, this approach has enabled more reliable resolution of repetitive AFP gene arrays [51], with similar benefits expected for complex NBS-LRR clusters in plants.
Phased assembly techniques: These methods distinguish between maternal and paternal haplotypes, reducing haplotype collapse and improving gene model accuracy in heterozygous individuals. Tools like hifiasm, Shasta, and Verkko generate graphical fragment assembly (GFA) files that represent assembly uncertainty, allowing researchers to assess confidence in problematic regions [51].
Error detection workflows: Specialized tools like gfa_parser (which computes and extracts all possible contiguous sequences from GFA files) and switch_error_screen (which flags potential phasing errors) help identify and mitigate assembly artifacts [51]. These are particularly valuable in repetitive regions where misassembly is common.
Evidence-based annotation: Integrating multiple lines of evidence significantly improves annotation accuracy. The MAKER pipeline combines ab initio predictions, homology evidence, and RNA-seq data to generate consensus gene models [56]. For optimal results, transcriptomic evidence should be assembled using tools like StringTie or Trinity and then incorporated into the annotation process [56].
Table 2: Key Bioinformatics Tools for Addressing Annotation Challenges
| Tool Category | Representative Tools | Primary Function | Application to NBS Gene Research |
|---|---|---|---|
| Genome Assembly | hifiasm, Verkko, Shasta | Long-read assembly and phasing | Resolving tandem NBS-LRR clusters |
| Gene Prediction | AUGUSTUS, BRAKER, Helixer | Ab initio gene finding | Initial identification of NBS domains |
| Evidence Integration | MAKER, EVidenceModeler | Combining diverse data sources | Improving NBS-LRR gene model accuracy |
| Pseudogene Detection | Custom pipelines [53] [54] | Identifying disablements | Filtering non-functional NBS sequences |
| Quality Assessment | BUSCO, OMArk, GeneValidator | Evaluating annotation completeness | Benchmarking NBS-LRR annotation quality |
Specialized Approaches for NBS-LRR Gene Annotation
The unique characteristics of NBS-LRR genes necessitate specialized annotation strategies. Based on successful implementations in multiple plant species [52] [57] [9], the following protocol is recommended:
Step 1: Domain-based identification
- Perform HMMER searches against the complete proteome using the NB-ARC domain (PF00931) from Pfam with e-value < 1e-04 [57]
- Confirm NBS domain presence using PfamScan with e-value < 1e-03
- Construct species-specific NBS HMM profile to increase sensitivity for divergent family members
Step 2: Structural classification
- Identify N-terminal domains (TIR, CC, RPW8) using Pfam and COILS [9]
- Classify genes into subclasses (TNL, CNL, RNL) based on domain architecture
- Identify truncated forms (NL, CN, N) that may represent pseudogenes or specialized variants
Step 3: Cluster identification and analysis
- Map gene locations to chromosomes or scaffolds where possible
- Identify tandem clusters (genes separated by ≤200 kb) [52]
- Analyze segmental duplications through whole-genome duplication detection
Step 4: Evolutionary analysis
- Perform all-versus-all BLAST to identify gene families (≥60% identity, ≥60% coverage) [9]
- Calculate nonsynonymous (Ka) and synonymous (Ks) substitution rates
- Identify positive selection using PAML site models (comparing M7 vs. M8)
This specialized approach has proven effective in multiple systems, such as the identification of 167 NBS-LRR genes in Dioscorea rotundata [52] and 1,015 in Malus domestica [57], providing reliable datasets for evolutionary inference.
Experimental Validation and Quality Control
Transcriptomic Validation Strategies
Experimental validation is crucial for verifying computational predictions and identifying functional genes. RNA-Seq data provides particularly valuable evidence for correcting gene models fragmented in the assembly process [50]. Recommended approaches include:
Multi-tissue transcriptomics: Sequencing RNA from multiple tissues and developmental stages helps verify gene models and identify constitutively expressed versus tissue-specific NBS-LRR genes. In Dioscorea rotundata, transcriptome analysis across four tissues revealed that "tuber and leaf displayed a relatively high NBS-LRR gene expression than the stem and flower" [52], providing insights into potential functional specialization.
Stress-induced expression profiling: Exposing plants to pathogens or elicitors and monitoring NBS-LRR expression helps identify functional resistance genes. In Fragaria species, expression profiling after pathogen infection showed that "the same gene expressed differently under different genetic backgrounds in response to pathogens" [9], highlighting the importance of genetic context.
Isoform sequencing: Long-read transcript sequencing (Iso-Seq) provides full-length transcript information that dramatically improves gene model accuracy, particularly for genes with multiple exons.
The following workflow diagram illustrates a comprehensive approach to gene annotation validation:
Quality Assessment Metrics
Rigorous quality assessment is essential for evaluating annotation reliability. Key metrics include:
BUSCO scores: Benchmarking Universal Single-Copy Orthologs assessments measure completeness based on evolutionarily conserved genes [56]. High BUSCO scores (≥90%) indicate comprehensive annotations, though they don't guarantee accuracy for lineage-specific families like NBS-LRR genes.
Orthology benchmark consistency: Comparing orthology inferences across different annotation sources helps identify systematic errors. Significant discrepancies in the "proportion of orthologous genes per genome" or the "completeness of Hierarchical Orthologous Groups" indicate annotation problems [55].
Synteny conservation: For NBS-LRR genes, examining syntenic relationships across related species can help validate gene models and identify evolutionary patterns. In Fragaria species, "shared hotspot regions of the duplicated NBS-LRRs on the chromosomes" provided evidence for lineage-specific duplications preceding species divergence [9].
Implications for NBS Gene Evolution Research
Impact on Evolutionary Inference
Accurate annotation is particularly crucial for understanding the evolutionary dynamics of NBS-LRR genes, which play critical roles in plant immunity and exhibit complex evolutionary patterns. Annotation errors can significantly distort key evolutionary inferences:
Gene birth-death rates: The NBS-LRR gene family follows a birth-death model with rapid turnover. Misannotation of pseudogenes as functional genes inflates birth rates, while missing functional genes due to assembly gaps deflates them. In the six Fragaria species, correctly identifying 1,134 NBS-LRR genes across 184 gene families enabled researchers to detect that "lineage-specific duplication of the NBS-LRR genes occurred before the divergence of the six Fragaria species" [9].
Selection pressure estimates: Proper classification of functional genes versus pseudogenes is essential for accurate calculation of Ka/Ks ratios. Studies have found that "the Ks and Ka/Ks ratios suggested that the TNLs are more rapidly evolving and driven by stronger diversifying selective pressures than the non-TNLs" [9], but these patterns could be obscured by annotation errors.
Evolutionary history reconstruction: Accurate orthology assignment is fundamental for understanding gene family evolution. Different annotation methods yield "markedly distinct orthology inferences" [55], which directly impact phylogenetic analyses and evolutionary conclusions about NBS-LRR gene evolution.
Best Practices for Evolutionary Studies
Based on current evidence, the following best practices are recommended for evolutionary studies of NBS-LRR genes:
Multi-genome consistency: When studying gene family evolution across multiple species, use consistent annotation methods rather than relying on published annotations generated with different pipelines. This reduces artifacts introduced by methodological differences [55].
Pseudogene-aware analyses: Explicitly identify and account for pseudogenes in evolutionary analyses. In the Rosaceae family, understanding NBS-LRR gene expansion required distinguishing functional genes from pseudogenes [57].
Expression-informed annotations: Incorporate transcriptomic data to validate gene models, particularly for fragmented assemblies where RNA-Seq can "connect genes that have been fragmented in the assembly process" [50].
Selective pressure analysis: Calculate Ka/Ks ratios separately for different NBS-LRR subclasses (TNLs vs. CNLs), as they may evolve under different selective constraints [9].
Table 3: Research Reagent Solutions for Annotation Validation
| Reagent/Resource | Primary Function | Application in NBS Gene Research |
|---|---|---|
| PacBio HiFi Reads | Long-read sequencing with high accuracy | Resolving complex NBS-LRR clusters |
| Hi-C Library Kits | Chromatin conformation capture | Scaffolding and phasing assemblies |
| RNA-Seq Library Prep Kits | Transcriptome sequencing | Validating gene models and expression |
| RACE Kits | Rapid amplification of cDNA ends | Verifying transcript start and end sites |
| Domain-Specific Antibodies | Protein detection | Confirming expression of NBS-LRR proteins |
| Pathogen Elicitors | Induction of defense responses | Testing functionality of R gene candidates |
Accurate gene annotation remains challenging but is essential for reliable evolutionary inference, particularly for rapidly evolving gene families like NBS-LRR genes. By implementing integrated approaches that combine high-quality genome assemblies with multiple lines of experimental evidence and sophisticated computational methods, researchers can significantly improve annotation accuracy. Specialized strategies for identifying pseudogenes, resolving complex gene clusters, and validating gene models through transcriptomics are particularly important for NBS-LRR gene research. As annotation methods continue to improve, so too will our understanding of the evolutionary dynamics that shape this critical gene family and its role in plant immunity.
In the study of plant disease resistance (R) genes, a significant proportion of nucleotide-binding site leucine-rich repeat (NBS-LRR) genes do not conform to the standard canonical domain architecture. Instead, they exhibit atypical or degenerated domains, presenting a substantial challenge for accurate gene classification and functional annotation. These non-canonical genes are not mere artifacts; they are often generated by local genome duplication events and can play crucial roles in the plant immune system, such as serving as adaptors or regulators in signaling pathways [58] [59].
The NBS-LRR gene family is the largest class of plant R genes, with approximately 80% of cloned R genes belonging to this family [60]. These genes are essential components of the plant's effector-triggered immunity (ETI) system, enabling plants to recognize specific pathogen effectors and initiate a robust immune response [61]. The canonical structure of these genes typically includes an N-terminal domain (TIR, CC, or RPW8), a central nucleotide-binding site (NBS) domain, and a C-terminal leucine-rich repeat (LRR) domain [59].
However, genome-wide studies across diverse plant species consistently reveal a abundance of genes that deviate from this typical architecture. For instance, in Salvia miltiorrhiza, among 196 NBS genes identified, only 62 possessed complete N-terminal and LRR domains [60]. Similarly, in Nicotiana benthamiana, 60 out of 156 NBS-LRR homologs were classified as N-type, containing only the NBS domain without typical N-terminal or LRR domains [59]. These atypical genes represent a significant portion of the NBS-LRR repertoire and require specialized approaches for accurate identification and classification.
The Challenge of Atypical Domain Architectures
Types and Prevalence of Domain Degeneration
Atypical NBS-LRR genes arise primarily through domain loss or sequence degeneration, resulting in distinct structural categories. Based on specific domain deletions, these atypical forms are classified into several subtypes:
- N-type (NBS only): Proteins containing only the NBS domain without N-terminal or LRR domains.
- CN-type (CC-NBS): Proteins with coiled-coil and NBS domains but lacking LRR domains.
- TN-type (TIR-NBS): Proteins with TIR and NBS domains but lacking LRR domains.
- NL-type (NBS-LRR): Proteins with NBS and LRR domains but lacking recognizable N-terminal domains [60] [59].
The prevalence of these degenerate forms varies significantly across plant species. The table below summarizes the distribution of atypical NBS genes across several recently studied plant species:
Table 1: Prevalence of Atypical NBS Genes in Various Plant Species
| Plant Species | Total NBS Genes Identified | Atypical NBS Genes | Most Prevalent Atypical Type | Reference |
|---|---|---|---|---|
| Salvia miltiorrhiza | 196 | 134 (68.4%) | CN-type (75 proteins) | [60] |
| Nicotiana benthamiana | 156 | 103 (66.0%) | N-type (60 proteins) | [59] |
| Dendrobium officinale | 74 | Not specified | Non-NBS-LRR subclass | [61] |
| Nicotiana tabacum | 603 | 329 (54.6%) | N-type (≈45.5% of total) | [7] |
Molecular Mechanisms Generating Atypical Genes
Local genome duplication events play a crucial role in generating atypical resistance genes. A seminal case study of the rice Pb1 gene demonstrates how genome duplication can create a functional atypical R gene. Pb1 encodes an atypical CC-NBS-LRR protein characterized by a apparently absent P-loop and other degenerated motifs in the NBS domain [58].
The Pb1 gene was located within one of tandemly repeated 60-kb units, which presumably arose through local genome duplication. This duplication event placed a promoter sequence upstream of a previously transcriptionally inactive 'sleeping' resistance gene, conferring a characteristic expression pattern that increases during plant development and accounts for adult/panicle resistance [58]. This mechanism highlights how gene duplication can generate new functional genes with atypical architectures.
Beyond creating new promoters, duplication events can lead to subfunctionalization or pseudogenization through partial gene duplication, resulting in truncated genes lacking complete domain sets [61] [62]. The XTNX gene family, which contains highly divergent TIR and NBS domains, represents another class of atypical resistance genes that originated in land plants and has undergone specific conservative evolution patterns [62].
Methodological Framework for Classification
Domain Identification and Verification
Accurate classification of atypical NBS-LRR genes begins with comprehensive domain identification. The following experimental protocol outlines a robust pipeline for domain identification and verification:
Table 2: Key Research Reagents and Tools for Domain Identification
| Reagent/Tool | Function | Key Features/Parameters |
|---|---|---|
| HMMER v3.1b2 | Hidden Markov Model search for conservative domains | Uses PF00931 (NB-ARC) model; E-value < 1×10⁻²⁰ [59] [7] |
| Pfam Database | Protein family database for domain annotation | Confirms NBS domain with E-values < 0.01 [59] |
| SMART Tool | Domain architecture analysis | Identifies specific domains and their boundaries [59] |
| NCBI CDD | Conserved Domain Database search | Verifies coiled-coil domains and domain completeness [7] |
| MEME Suite | Motif discovery and analysis | Identifies conserved motifs; parameters: motif count 10, width 6-50 aa [59] |
Experimental Protocol 1: Domain Identification Pipeline
- Initial Domain Screening: Perform HMMER search against the target genome or protein dataset using the NB-ARC domain model (PF00931) with an E-value cutoff of 1×10⁻²⁰ to identify candidate NBS-containing genes [59] [7].
- Domain Verification: Submit candidate sequences to the Pfam database to confirm the complete presence of the NBS domain with E-values below 0.01 [59].
- Comprehensive Domain Annotation: Use multiple tools (SMART, NCBI CDD, and InterProScan) to identify all potential domains, including TIR, CC, RPW8, and LRR domains [59] [63].
- Motif Analysis: Conduct motif analysis using MEME with default parameters to identify conserved motifs beyond the core domains [59].
- Manual Curation: Manually verify domain boundaries and architecture, particularly for degenerate domains that may not be recognized by standard tools [58].
This multi-tool approach is essential because atypical domains often exhibit significant sequence divergence that may not be recognized by a single method. For example, the NBS domain in XTNX proteins is only half the length of a regular NBS domain and is often annotated as AAA or P-loop superfamily domains by standard tools [62].
Figure 1: Workflow for Domain Identification and Verification
Phylogenetic and Structural Analysis
Once domains are identified, phylogenetic and structural analyses provide critical context for classifying atypical genes:
Experimental Protocol 2: Phylogenetic Classification
- Sequence Alignment: Perform multiple sequence alignment of NBS-LRR protein sequences using MUSCLE v3.8.31 or Clustal W with default parameters [59] [7].
- Phylogenetic Reconstruction: Construct phylogenetic trees using Maximum Likelihood method in MEGA11 or similar software with 1000 bootstrap replicates to assess node support [61] [59].
- Classification: Classify sequences into clades (CNL, TNL, RNL) and identify atypical genes that fall outside these main clades or form distinct lineages [60] [61].
- Structural Analysis: Analyze gene structure using annotation files (GFF3) to visualize exon-intron patterns, which often differ between typical and atypical NBS-LRR genes [59].
Phylogenetic analysis of CNL-type proteins across multiple plant species has revealed that orchid NBS-LRR genes have significantly degenerated on specific phylogenetic branches, providing evolutionary context for domain loss patterns [61]. Similarly, analysis of XTNX genes shows they form distinct clades separate from typical TNL genes, supporting their classification as a unique gene family with a different evolutionary origin [62].
Advanced Computational Approaches
Machine Learning and Deep Learning Classification
Traditional similarity-based methods often fail to identify atypical R genes due to low sequence homology. To address this limitation, advanced computational approaches have been developed:
PRGminer represents a cutting-edge deep learning-based tool specifically designed for accurate prediction of resistance proteins, including those with atypical architectures [63]. The tool operates in two phases:
- Phase I: Predicts input protein sequences as R-genes or non-R-genes using dipeptide composition features, achieving 98.75% accuracy in k-fold testing.
- Phase II: Classifies predicted R-genes into eight different classes (CNL, TNL, RNL, Kinase, RLP, LECRK, RLK, LYK) with 97.55% overall accuracy [63].
The superior performance of deep learning approaches stems from their ability to extract higher-level features from raw encoded protein sequences based on classification rather than relying solely on traditional alignment-based methods [63].
Figure 2: PRGminer Two-Phase Deep Learning Workflow
Genomic Context and Synteny Analysis
Analyzing genomic context provides another powerful approach for identifying and classifying atypical R genes:
Experimental Protocol 3: Genomic Context Analysis
- Tandem Duplication Detection: Use MCScanX or similar tools to identify tandemly duplicated genes, which are common for R-genes [7].
- Synteny Analysis: Perform syntenic analysis across related species to identify conserved R-gene clusters and detect species-specific gains or losses [7].
- Selection Pressure Analysis: Calculate non-synonymous (Ka) and synonymous (Ks) substitution rates using KaKs_Calculator to identify genes under positive selection, which often indicates functional importance [7].
NBS-LRR genes are often organized in clusters of closely duplicated genes, though they may also exist as individual units scattered across the genome [63]. The presence of atypical genes within these clusters provides important evolutionary context. For example, in Nicotiana tabacum, 76.62% of NBS members could be traced back to their parental genomes, demonstrating the conservation of these genes after polyploidization [7].
Functional Validation of Atypical Genes
Expression Analysis
Transcriptome analysis under various conditions provides crucial evidence for the functional relevance of atypical R genes:
Experimental Protocol 4: Expression Profiling
- RNA-Seq Data Collection: Download and quality-check RNA-seq datasets from relevant experiments (e.g., pathogen infection, hormone treatment) [61] [7].
- Read Mapping and Quantification: Map cleaned sequencing reads to the reference genome using Hisat2 and perform transcript quantification with Cufflinks or similar tools [7].
- Differential Expression Analysis: Identify differentially expressed genes (DEGs) using Cuffdiff or DESeq2, focusing on NBS-LRR genes showing significant expression changes [61] [7].
- Co-expression Network Analysis: Perform weighted gene co-expression network analysis (WGCNA) to identify clusters of co-expressed genes and correlate them with specific pathways [61].
For example, in Dendrobium officinale, transcriptome analysis following salicylic acid (SA) treatment identified 1,677 differentially expressed genes, including six NBS-LRR genes that were significantly up-regulated [61]. One of these genes (Dof020138) showed close association with pathogen identification pathways, MAPK signaling pathways, and plant hormone signal transduction pathways, suggesting its importance in the immune response [61].
Promoter and Cis-Element Analysis
Promoter analysis can reveal important clues about the regulation and potential functions of atypical R genes:
Experimental Protocol 5: Promoter Analysis
- Promoter Sequence Extraction: Extract 1500 bp upstream sequences from the translation start site [59].
- Cis-Element Identification: Use PlantCARE or similar databases to identify cis-acting regulatory elements in the promoter regions [59].
- Element Classification: Categorize identified elements into functional groups (hormone-responsive, stress-responsive, development-related) [59].
- Expression Correlation: Correlate specific cis-elements with expression patterns under different conditions [60].
In Salvia miltiorrhiza, promoter analysis demonstrated an abundance of cis-acting elements in SmNBS genes related to plant hormones and abiotic stress, providing insights into their potential regulatory mechanisms [60]. Similarly, in Nicotiana benthamiana, promoter analysis of NBS-LRR genes detected 29 shared kinds of cis-elements and 4 kinds unique to irregular-type NBS-LRR genes, indicating potential differences in their upstream regulation [59].
The classification of genes with atypical or degenerated domains represents a significant challenge in plant genomics, particularly in the context of NBS-LRR gene evolution. The framework presented in this whitepaper—integrating comprehensive domain identification, phylogenetic analysis, advanced computational methods, and functional validation—provides a systematic approach for addressing this complexity.
Gene duplication events emerge as a central mechanism generating architectural diversity in NBS-LRR genes, from local tandem duplications creating novel promoters for sleeping genes to whole-genome duplications facilitating subfunctionalization of duplicated copies. The case study of the rice Pb1 gene exemplifies how local genome duplication can generate functional atypical R genes through promoter acquisition [58].
As genomic data continue to accumulate, the integration of deep learning approaches like PRGminer with traditional comparative genomic methods will become increasingly powerful for identifying and classifying these challenging genes [63]. This integrated approach is essential for fully understanding the evolutionary dynamics of plant immune genes and harnessing their diversity for crop improvement strategies.
Gene duplication is a fundamental evolutionary process that provides the raw material for functional innovation, yet it creates a significant challenge for researchers: functional redundancy. Dense gene clusters, particularly those arising from tandem duplication events, are hotspots for genetic innovation but complicate the identification of which specific genes merit prioritization for functional characterization. This challenge is acutely present in the study of Nucleotide-binding site Leucine-rich repeat (NBS-LRR) genes, which are crucial for plant disease resistance and exhibit remarkable proliferation in plant genomes through various duplication mechanisms [64] [65].
The very nature of duplication events creates families of similar genes where functional redundancy can mask phenotypic effects when individual genes are disrupted. In Arabidopsis thaliana, for instance, the NBS-LRR gene family shows higher-than-average levels of structural divergence following duplication, suggesting these genes are under selection for rapid evolution of gene structure [65]. Similarly, studies in Aurantioideae species reveal that tandem and proximal duplication types undergo rapid functional divergence, as evidenced by their evolutionary rates [66]. This whitepaper synthesizes current methodologies and frameworks to help researchers navigate this complexity, providing a systematic approach for prioritizing candidate genes within dense clusters, with special emphasis on NBS gene evolution.
The Evolutionary Context of Gene Duplication
Modes of Gene Duplication and Their Consequences
Gene duplication occurs through several distinct mechanisms, each with different implications for gene function and evolution. Understanding these modes is essential for interpreting cluster architecture and potential functional relationships. Research across plant genomes has identified five primary duplication types: Whole-genome duplication (WGD), tandem duplication (TD), proximal duplication (PD), transposed duplication (TRD), and dispersed duplication (DSD) [66].
Table 1: Modes of Gene Duplication and Their Characteristics
| Duplication Type | Mechanism | Typical Cluster Size | Structural Divergence | Prevalent in NBS Genes |
|---|---|---|---|---|
| Whole-genome duplication (WGD) | Complete genome copying | Variable, often genome-wide | Lower initial divergence | Yes, but often followed by fractionation |
| Tandem duplication (TD) | Unequal crossing over | Small to large clusters | Moderate to high | Yes, very prevalent |
| Proximal duplication (PD) | Regional duplication mechanisms | Small clusters | Moderate | Yes |
| Transposed duplication (TRD) | DNA or RNA-mediated transposition | Often single genes | High, often biased | Yes |
| Dispersed duplication (DSD) | Various mechanisms including transposition | Single genes | Variable | Less common |
Different duplication modes leave distinct genomic signatures and exhibit varying rates of sequence and structural evolution. Transposed duplicates, for instance, show the most dramatic structural divergence, with parental loci typically having longer coding regions and exons, while transposed loci accumulate more insertions and deletions [65]. In the Aurantioideae subfamily, which includes citrus species, tandem duplication is the predominant duplication type, confirming its importance in genome evolution and expansion [66].
Evolutionary Forces Acting on Duplicated Genes
Following duplication, genes experience various evolutionary forces that determine their fate. The Ka/Ks ratio (non-synonymous to synonymous substitution rate) serves as a key indicator of selective pressure, with values <1 indicating purifying selection, ≈1 suggesting neutral evolution, and >1 implying positive selection [66]. In barley genomes, genes involved in evolutionary "arms races" – particularly pathogen defence genes – show statistical associations with duplication-prone regions, highlighting how selective pressures shape cluster evolution [14].
NBS-LRR genes exemplify how antagonistic co-evolution with pathogens drives gene family expansion and diversification. These genes are among the most variable gene families in plants, likely due to pathogen-driven selection pressures [64]. The continuous co-evolution of genetic elements through intragenomic conflict or host-pathogen conflict creates a molecular "arms race" that maintains genetic diversity within clusters [67].
Analytical Frameworks for Prioritization
Genomic and Phylogenetic Approaches
Comparative genomics provides powerful tools for identifying evolutionarily significant genes within clusters. By examining orthologous relationships across related species, researchers can pinpoint conserved genes that may retain critical functions. In Asparagus species, for instance, comparative analysis of NLR genes across A. officinalis, A. kiusianus, and A. setaceus revealed a marked contraction of the NLR gene repertoire during domestication, with only 16 conserved NLR gene pairs maintained between wild and domesticated species [64]. Such conserved genes represent prime candidates for functional analysis.
Phylogenetic reconstruction coupled with domain architecture analysis enables the classification of NBS-LRR genes into distinct subfamilies (CNLs, TNLs, and RNLs) based on their N-terminal domains [64]. This classification provides a framework for assessing functional diversity within clusters. Maximum likelihood methods implemented in tools like MEGA can establish evolutionary relationships, while domain analysis using InterProScan and NCBI's Batch CD-Search confirms protein architectures [64].
Table 2: Genomic Approaches for Gene Prioritization
| Method | Application | Tools/Implementation | Interpretation |
|---|---|---|---|
| Ortholog Analysis | Identify evolutionarily conserved genes | OrthoFinder, BLAST | Conserved genes across species may have essential functions |
| Ka/Ks Calculation | Detect selection pressure | Code in R/Python, KAKS_calculator | Ka/Ks >1 indicates positive selection; <1 indicates purifying selection |
| Domain Architecture Analysis | Classify genes into functional subtypes | InterProScan, NCBI CD-Search | Different domain combinations suggest functional specialization |
| Cluster Pattern Analysis | Identify recent expansions | MCScanX, BEDTools | Recent expansions may indicate response to pathogen pressures |
| Structural Variation Analysis | Detect presence/absence variations | SV callers (Delly, Lumpy) | Presence/absence variations may correlate with phenotypic differences |
Expression-Based Prioritization Strategies
Gene expression profiling provides critical insights into functional differentiation among duplicated genes. Several analytical approaches can extract meaningful patterns from expression data:
Cluster analysis of large-scale gene expression data from time-course experiments can reveal correlated expression patterns that conform to shared pathways and control processes [68]. This approach leverages algorithms to group genes with similar expression profiles, suggesting co-regulation or functional relationships.
Comparative expression analysis between different tissues, developmental stages, or stress conditions can identify genes with specialized expression patterns. In Aurantioideae, for example, comparing gene expression differentiation between outer and inner pericarps of Citrus maxima revealed that the proportion of differentiated expression was generally higher in the exocarp, suggesting tissue-specific functional roles for duplicated genes [66].
When analyzing expression data, proper data normalization is essential, as variables with different scales can dominate the clustering process if not properly standardized [69]. Additionally, dimensionality reduction techniques such as principal component analysis (PCA) or t-SNE can help visualize complex expression relationships in lower-dimensional space [69].
Emerging AI-Based Approaches
Generative genomic models represent a cutting-edge approach for function-guided gene design and prioritization. The Evo model, a genomic language model trained on prokaryotic DNA sequences, can leverage genomic context to perform "semantic design" – generating novel sequences enriched for targeted biological functions based on their association with known functional genes [70].
This approach effectively operationalizes the "guilt by association" principle at scale, using a model's understanding of multi-gene relationships in prokaryotic genomes to identify genes likely to share functions based on their genomic neighborhood [70]. While developed for prokaryotic systems, this conceptual framework holds promise for prioritizing genes in plant NBS clusters based on their genomic context and association with characterized resistance genes.
Experimental Validation Workflows
A Framework for Functional Characterization
Prioritized candidate genes require rigorous experimental validation to confirm their functions. The following workflow outlines a systematic approach for characterizing NBS-LRR genes:
Diagram 1: Gene Validation Workflow
This workflow begins with detailed expression analysis using qRT-PCR or RNA-seq to verify expression patterns under relevant conditions. In Asparagus NLR studies, most preserved NLR genes in domesticated A. officinalis showed either unchanged or downregulated expression following fungal challenge, indicating potential functional impairment in disease resistance mechanisms [64].
Subcellular localization studies using tools like WoLF PSORT can predict and confirm protein localization [64], while protein interaction screening through yeast-two-hybrid (Y2H) or co-immunoprecipitation (Co-IP) assays can identify signaling partners. Functional genetic approaches, particularly CRISPR-Cas9 mediated knockout or overexpression, can establish gene-phenotype relationships, followed by comprehensive phenotypic assays to quantify disease response outcomes.
High-Throughput Functional Screening
For dense gene clusters where multiple candidates exist, high-throughput screening methods can accelerate functional characterization:
Heterologous expression systems enable rapid testing of gene function in model systems. For NBS-LRR genes, this might involve expressing candidate genes in susceptible plant varieties and challenging with relevant pathogens.
Virus-induced gene silencing (VIGS) provides an efficient approach for transient gene knockdown, allowing rapid assessment of gene function without stable transformation.
Multiplexed CRISPR approaches now enable simultaneous targeting of multiple gene family members, helping overcome functional redundancy by creating higher-order mutants.
Table 3: Research Reagent Solutions for Gene Cluster Analysis
| Reagent/Resource | Function | Application Example |
|---|---|---|
| PlantCARE Database | Identification of cis-acting regulatory elements | Analysis of 2000bp promoter sequences upstream of ATG codon [64] |
| InterProScan | Protein domain classification and functional analysis | Characterizing NBS, TIR, CC, and LRR domains in NLR proteins [64] |
| MEME Suite | Discovery of conserved protein motifs | Identifying conserved motifs within NBS domains [64] |
| OrthoFinder | Clustering of orthologous genes across species | Identifying conserved NLR gene pairs between species [64] |
| SynGenome Database | Access to AI-generated genomic sequences | Exploring semantic design for functional gene discovery [70] |
| PRGdb 4.0 | Plant resistance gene database | Classification and comparison of NLR genes [64] |
| WoLF PSORT | Prediction of protein subcellular localization | Determining NLR protein localization [64] |
| PlantGARDEN | Genomic resource repository | Accessing genomic data for comparative analyses (e.g., A. kiusianus) [64] |
Case Studies and Applications
NLR Gene Family in Asparagus Species
A comprehensive analysis of NLR genes across three Asparagus species (A. officinalis, A. kiusianus, and A. setaceus) demonstrates practical application of prioritization approaches. Researchers identified 63, 47, and 27 NLR genes in A. setaceus, A. kiusianus, and A. officinalis, respectively, revealing a marked contraction during domestication [64]. Orthologous analysis identified 16 conserved NLR gene pairs between A. setaceus and A. officinalis, which likely represent the NLR genes preserved during domestication and thus prime candidates for essential immune functions [64].
Expression profiling following Phomopsis asparagi infection showed distinct patterns: while A. setaceus remained asymptomatic, A. officinalis was susceptible, and most preserved NLR genes in A. officinalis showed either unchanged or downregulated expression after fungal challenge [64]. This integrated approach – combining comparative genomics, evolutionary analysis, and expression profiling – successfully identified candidate genes potentially responsible for differential disease susceptibility.
Semantic Design for Novel Gene Discovery
The Evo genomic language model demonstrates how AI approaches can extend beyond natural sequence variation. Researchers applied "semantic design" to generate novel anti-CRISPR proteins and toxin-antitoxin systems, including de novo genes with no significant sequence similarity to natural proteins [70]. This approach achieved robust activity and high experimental success rates even without structural priors or known evolutionary conservation [70].
For NBS gene researchers, this methodology suggests a path for exploring novel resistance genes beyond natural variation. By leveraging the genomic context of known functional NBS-LRR genes, researchers could potentially generate synthetic resistance genes with enhanced or novel recognition capabilities.
Overcoming functional redundancy in dense gene clusters requires a multi-faceted approach that combines evolutionary analysis, comparative genomics, expression profiling, and emerging computational methods. No single methodology suffices; rather, prioritization is most effective when multiple lines of evidence converge on candidate genes.
For NBS-LRR genes specifically, the evolutionary context of duplication events provides critical insights – genes in rapidly expanding clusters with signatures of positive selection may represent recent adaptations to pathogen pressure, while evolutionarily conserved genes may encode core immune functions. As genomic technologies advance, particularly in AI-based sequence design and single-cell expression profiling, researchers will gain increasingly powerful tools to navigate the complexity of dense gene clusters and unlock their functional secrets.
The strategies outlined herein provide a framework for systematically prioritizing candidate genes, accelerating the translation of genomic data into biological understanding and, ultimately, improved crop varieties with enhanced disease resistance.
In plant genomes, genes involved in arms races, such as those encoding nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins, exhibit remarkable dynamism and expansion. Tandem duplication serves as a key mechanism for rapidly generating this crucial genetic diversity, allowing organisms to adapt to evolving pathogenic threats [14]. Genes located within duplication-prone genomic regions, particularly those rich in long tandem repeats, can more freely explore mutational space, leading to the efficient generation of novel resistance specificities [14]. This evolutionary process results in a measurable statistical association between arms-race genes and duplication-inducing elements, supporting a model of effective cooperation between selfish replicators and the genes they duplicate [14].
The accurate delineation of recently expanded gene families, especially those with tandemly duplicated architectures, presents significant technical challenges. These include resolving highly similar paralogous sequences, distinguishing functional genes from pseudogenes, and accurately quantifying copy number variations across complex genomic regions. This technical guide provides a comprehensive framework for optimizing tandem repeat analysis, with specific application to the study of NBS gene evolution. By integrating state-of-the-art bioinformatic tools, evolutionary analyses, and functional validation methods, researchers can overcome these challenges to gain novel insights into plant immunity and adaptive evolution.
Genomic Landscape of Tandemly Repeated NBS Genes
Evolutionary Patterns in Plant Genomes
Recent pan-genomic studies in maize have revealed extensive presence-absence variation (PAV) within the ZmNBS gene family, distinguishing conserved "core" subgroups (e.g., ZmNBS31, ZmNBS17-19) from highly variable "adaptive" subgroups (e.g., ZmNBS1-10, ZmNBS43-60) [11]. This core-adaptive model of resistance gene evolution provides a conceptual framework for understanding how duplication mechanisms and selection pressures jointly shape the evolution of disease resistance genes.
Different duplication mechanisms show distinct evolutionary preferences: canonical CNL/CN genes largely originate from dispersed duplications, while N-type genes are enriched in tandem duplications [11]. Evolutionary rate analysis further demonstrates that whole-genome duplication (WGD)-derived genes experience strong purifying selection (low Ka/Ks), whereas tandem and proximal duplications (TD/PD) show signs of relaxed or positive selection, enabling greater functional diversification [11].
Association with Duplication-Prone Genomic Regions
In barley, sophisticated genomic analyses have confirmed that natural selection has favored lineages in which arms-race genes—particularly pathogen defence genes—are associated with duplication-inducers, most notably Kb-scale tandem repeats [14]. These duplication-prone regions show a history of repeated long-distance 'dispersal' to distant genomic sites, followed by local expansion by tandem duplication [14].
Table 1: Genomic Tools for Tandem Repeat and NBS Gene Analysis
| Tool Name | Primary Function | Application in NBS Gene Analysis | Reference |
|---|---|---|---|
| RepeatsDB | Classification/annotation of structured tandem repeat proteins (STRPs) | Annotation of tandem repeat domains in NBS proteins | [71] |
| MCScanX | Analysis of segmental and tandem duplication events | Identifying tandemly duplicated NBS genes across genomes | [7] |
| STRchive | Clinical annotation of short tandem repeats (STRs) | Pathogenicity assessment of expanded repeat regions | [72] |
| PROST | Identification of spatially variable genes | Analyzing spatial expression patterns of expanded gene families | [73] |
| HMMER (PF00931) | Domain-based identification of NBS-LRR genes | Comprehensive identification of NBS family members | [7] |
Methodological Framework for Delineating Expanded Gene Families
Comprehensive Identification and Classification
The accurate identification of NBS-LRR genes across plant genomes requires a multi-step domain-based approach. The foundational step involves hidden Markov model (HMM) searches using the PF00931 (NB-ARC) model from the PFAM database to identify core NBS domains [7]. Subsequent domain characterization should include:
- TIR and LRR domains: Identified using multiple PFAM domains (PF01582, PF00560, PF07723, PF07725, PF12779, PF13306, PF13516, PF13855, PF14580, PF03382, PF01030, PF05725)
- Coiled-coil (CC) domains: Confirmed through the NCBI Conserved Domain Database (CDD)
- Completeness verification: All putative domains should be validated through NCBI's conserved domain functionality [7]
This systematic approach enabled the identification of 1,226 NBS genes across three Nicotiana genomes, revealing that approximately 45.5% contained only the NBS domain, while 23.3% were CC-NBS type, and only 2.5% were TIR-NBS members [7].
Phylogenetic and Evolutionary Analysis
Multiple sequence alignment of identified NBS-LRR protein sequences should be performed using tools such as MUSCLE with default parameters [7]. Phylogenetic reconstruction can then be conducted using MEGA11 with neighbor-joining methods and bootstrap validation (1,000 replicates). For evolutionary analysis, the following pipeline is recommended:
- Determine duplication modes: Analyze whole-genome duplication using self-BLASTP, then identify segmental and tandem duplication events with MCScanX [7]
- Establish syntenic relationships: Identify syntenic blocks across genomes through reciprocal BLASTP searches followed by MCScanX-based collinearity detection
- Calculate selection pressures: Process syntenic gene pairs with ParaAT and calculate non-synonymous (Ka) and synonymous (Ks) substitution rates using KaKs_Calculator 2.0 with appropriate evolutionary models [7]
This approach successfully demonstrated that in Fragaria species, TNLs exhibit significantly higher Ks and Ka/Ks values than non-TNLs, indicating more rapid evolution under stronger diversifying selection pressures [74].
Diagram 1: Bioinformatics workflow for NBS gene identification and evolutionary analysis. The pipeline begins with domain identification and progresses through phylogenetic reconstruction and selection pressure analysis.
Advanced Techniques for Tandem Repeat Analysis
Detection of Duplication-Prone Genomic Regions
Innovative approaches have been developed to identify Long-Duplication-Prone Regions (LDPRs) through scanning genome self-alignments for intervals with elevated amounts of locally-repeated sequences in the Kbp-scale length range [14]. This gene-agnostic method involves:
- Genome self-alignment: Using tools like BLAST or minimap2 to identify repeated sequences
- Interval analysis: Scanning for genomic regions with dense clusters of local duplications
- Threshold application: Defining LDPRs based on statistical thresholds for duplication density
- Gene association mapping: Identifying gene clusters statistically over-represented in LDPRs
Application in barley revealed 1,199 candidate LDPRs with lengths ranging between 5.5 and 1,123.598 Kbp (median length 33.600 Kbp), located primarily in subtelomeric regions of all chromosomes [14].
Spatial Expression Analysis
The PROST framework provides advanced capabilities for identifying spatially variable genes (SVGs) through the PROST Index, which quantitatively characterizes spatial gene expression patterns without statistical hypothesis testing [73]. The PROST workflow includes:
- Grid transformation: Interpolating irregular spots to regular grids using gene expressions with spatial locations
- Image processing: Applying min-max normalization and Gaussian filtering
- Region classification: Dividing gene grid images into foreground and background sub-regions
- Index calculation: Computing PROST Index scores based on Significance and Separability factors
This approach enables unsupervised clustering of spatial domains through a self-attention mechanism that integrates spatial information and gene expressions, significantly improving domain segmentation accuracy as measured by Adjusted Rand Index (ARI) and Normalized Mutual Information (NMI) metrics [73].
Short Tandem Repeat Analysis in Clinical Contexts
For clinical applications, VarSeq combined with STRchive annotations provides a streamlined workflow for analyzing and reporting short tandem repeats (STRs) alongside small variants [72]. The methodology includes:
- STR calling: Using specialized pipelines (DRAGEN, PacBio, Oxford Nanopore) for detection
- Filtering workflow: Applying filters for repeat unit validity and overlap with STRchive annotations
- Pathogenicity assessment: Leveraging STRchive database for clinical interpretations
- Report generation: Creating comprehensive clinical reports with repeat counts, associated diseases, and classification criteria
Large-scale validation using 9,580 exomes demonstrated that ES-based STR analysis identified pathogenic expansions in 0.6% of cases, with 0.3% receiving explanatory diagnoses from STR findings [75].
Table 2: Selection Pressure Patterns in Duplicated NBS Genes
| Duplication Mechanism | Evolutionary Pattern | Ka/Ks Signature | Functional Implications |
|---|---|---|---|
| Whole-Genome Duplication (WGD) | Strong purifying selection | Low Ka/Ks | Conservation of core immune functions |
| Tandem & Proximal Duplication (TD/PD) | Relaxed/positive selection | Higher Ka/Ks | Functional diversification and neofunctionalization |
| TIR-NBS-LRR (TNL) genes | Rapid evolution under diversifying selection | Higher Ks and Ka/Ks | Enhanced pathogen recognition specificity |
| Non-TNL genes | Slower evolution under stabilizing selection | Lower Ks and Ka/Ks | Maintenance of conserved signaling modules |
Experimental Protocols for Validation and Functional Characterization
Gene Expression Analysis Under Pathogen Challenge
RNA-seq analysis provides critical functional validation for expanded NBS gene families. The recommended protocol includes:
- Data acquisition: Download RNA-seq datasets from NCBI SRA (e.g., SRP310543 for black shank resistance in Nicotiana tabacum)
- Quality control: Convert SRA to FASTQ format using fastq-dump v2.6.3, followed by quality trimming with Trimmomatic v0.36 (minimum read length: 90 bp)
- Read mapping: Align cleaned reads to reference genome using Hisat2
- Expression quantification: Perform transcript quantification with Cufflinks v2.2.1 using FPKM normalization
- Differential expression: Identify differentially expressed genes through Cuffdiff [7]
This approach successfully revealed that the same NBS-LRR gene can express differently under various genetic backgrounds in response to pathogens, highlighting the functional plasticity of expanded gene families [74].
Functional Validation Through Genetic Manipulation
Several methods enable functional characterization of tandemly expanded NBS genes:
- Gene silencing: Using VIGS (Virus-Induced Gene Silencing) to assess resistance reduction, as demonstrated by decreased Verticillium dahliae resistance in cotton upon NBS-LRR silencing [7]
- Heterologous expression: Transforming Arabidopsis thaliana with maize NBS-LRR genes to evaluate improved resistance to Pseudomonas syringae [7]
- Overexpression studies: Conferring broad-spectrum resistance to viral pathogens through soybean TNL gene overexpression [7]
- Genome editing: Utilizing efficient CRISPR/Cas9 systems with novel promoters achieving near 100% homozygous mutation rates in dicotyledonous plants [7]
Research Reagent Solutions for Tandem Repeat Analysis
Table 3: Essential Research Reagents and Tools for Tandem Repeat Studies
| Reagent/Tool | Specific Application | Function/Benefit | Implementation Example |
|---|---|---|---|
| HMMER v3.1b2 with PF00931 | NBS domain identification | Foundation for comprehensive NBS-LRR family member identification | Identified 1,226 NBS genes across three Nicotiana genomes [7] |
| STRchive database | Pathogenicity annotation of STRs | Provides clinical interpretations and disease associations for expanded repeats | Enabled identification of pathogenic expansions in 0.6% of clinical exomes [75] |
| KaKs_Calculator 2.0 | Selection pressure analysis | Quantifies evolutionary pressures on duplicated genes | Revealed TNLs evolve faster than non-TNLs in Fragaria [74] |
| PROST Algorithm | Spatial expression analysis | Quantifies spatial gene expression patterns without statistical assumptions | Superior performance in spatial domain identification (ARI metrics) [73] |
| RepeatsDB | Structured tandem repeat protein annotation | Classifies and annotates STRPs from PDB and AlphaFoldDB | Expanded annotations for >34,000 unique protein sequences [71] |
The integration of advanced bioinformatic tools, evolutionary analyses, and functional validation methods provides a powerful framework for delineating recently expanded gene families. The association between arms-race genes and duplication-inducing elements represents a fundamental evolutionary strategy for generating diversity in plant immune systems [14]. Future methodologies will likely focus on single-cell spatial transcriptomics to resolve expression patterns at cellular resolution, long-read sequencing to completely resolve complex tandem arrays, and machine learning approaches to predict expansion-associated functional innovations.
As sequencing technologies continue to advance and multi-omics integration becomes more sophisticated, our ability to accurately delineate and functionally characterize expanded gene families will dramatically improve. This progress will not only deepen our understanding of plant-pathogen coevolution but also facilitate the identification of superior resistance gene candidates for crop improvement strategies. The systematic approach outlined in this guide provides a foundation for these future advances in tandem repeat analysis and NBS gene evolution research.
Beyond Prediction: Experimental Validation and Cross-Species Comparative Genomics
Functional genomics relies on robust techniques to validate gene function, particularly in the study of evolutionarily dynamic gene families. Virus-Induced Gene Silencing (VIGS) and transgenic complementation represent powerful reverse genetics approaches for confirming gene-phenotype relationships. Within plant immunity research, these methods are indispensable for characterizing nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes, which exhibit remarkable diversification through gene duplication events. This technical guide examines the mechanistic basis, experimental protocols, and applications of VIGS and complementation, providing a framework for their implementation in studying gene family evolution.
Theoretical Foundations and Mechanisms
Virus-Induced Gene Silencing (VIGS)
VIGS is an RNA-mediated reverse genetics technique that exploits the plant's innate antiviral defense mechanism to silence endogenous genes. The process involves post-transcriptional gene silencing (PTGS), an epigenetic phenomenon that results in sequence-specific degradation of target mRNAs [76]. When plants detect invasive viral transcripts, they activate a conserved defense pathway that ultimately degrees both viral and homologous host RNAs.
The molecular mechanism of VIGS unfolds through a defined series of events, visualized in Figure 1 and detailed below:
Figure 1: Mechanism of Virus-Induced Gene Silencing (VIGS)
Vector Delivery & Transcription: Recombinant viral vectors containing 300-500 bp fragments of the target gene are introduced into plant cells, typically via Agrobacterium-mediated transformation. The T-DNA containing the viral genome is transcribed into single-stranded RNA (ssRNA) within the host [77].
dsRNA Formation: Host RNA-dependent RNA polymerase (RdRP) recognizes viral ssRNA and synthesizes complementary strands, forming double-stranded RNA (dsRNA) molecules [76].
siRNA Generation: Dicer-like enzymes recognize and cleave dsRNA into short interfering RNA (siRNA) duplexes of 21-24 nucleotides [76].
RISC Assembly: siRNAs are incorporated into the RNA-induced silencing complex (RISC), which uses the siRNA as a guide to identify complementary mRNA sequences [76] [77].
Target Degradation: RISC specifically cleaves endogenous mRNAs complementary to the siRNA guide, resulting in post-transcriptional silencing of the target gene [77].
Beyond cytoplasmic mRNA degradation, VIGS can induce heritable epigenetic modifications through RNA-directed DNA methylation (RdDM). When siRNAs enter the nucleus, they can guide DNA methyltransferases to homologous genomic sequences, establishing stable transcriptional gene silencing that may persist across generations [76].
Transgenic Complementation
Transgenic complementation represents the reciprocal approach to VIGS, functioning to confirm gene function through restoration of phenotype via introduction of a functional gene copy. This methodology follows a "loss-of-function, gain-of-function" logical framework, providing compelling evidence for gene identity.
Figure 2: Transgenic Complementation Logic Flow
The power of transgenic complementation lies in its ability to provide direct evidence that a specific gene is responsible for a particular phenotype. When a mutant phenotype is reversed by introducing a wild-type version of the candidate gene, it establishes a causal relationship rather than merely correlation.
Technical Protocols and Methodologies
VIGS Experimental Workflow
Implementing VIGS requires careful execution of sequential steps, from vector design to phenotypic analysis. The complete workflow is visualized in Figure 3, with detailed methodology following.
Figure 3: VIGS Experimental Workflow
Vector Selection and Construction
The choice of viral vector depends on the host plant species and research objectives:
- Tobacco Rattle Virus (TRV): Widely used for broad host range (Solanaceae, Cruciferae, Gramineae) and efficient meristem invasion [77].
- Foxtail Mosaic Virus (FoMV): Particularly valuable for monocots including switchgrass, maize, and barley [78].
- Barley Stripe Mosaic Virus (BSMV): Effective in cereals and capable of silencing in roots and leaves [78].
For vector construction, a 300-500 bp fragment of the target gene is amplified and cloned into the viral vector. Critical considerations include:
- Avoid regions of high sequence similarity to non-target genes
- Exclude homopolymeric sequences that may cause recombination
- Select fragments with moderate GC content (40-60%)
- Verify insert orientation and sequence fidelity [77] [78]
Modern vectors often incorporate Gateway recombination sites or ligation-independent cloning (LIC) cassettes to facilitate rapid cloning [77].
Plant Inoculation Methods
Agroinfiltration Agrobacterium tumefaciens strains GV3101 or LBA4404 harboring the binary VIGS vector are grown in LB medium with appropriate antibiotics to OD₆₀₀ = 0.4-1.0. Cells are pelleted and resuspended in infiltration medium (10 mM MgCl₂, 10 mM MES, 150 μM acetosyringone). The abaxial surface of leaves is infiltrated using a needleless syringe, applying gentle pressure until the infiltration zone becomes water-soaked [77] [78].
Rub-Inoculation Virus-containing sap is prepared by grinding 4g of infected leaf tissue in 16ml of potassium phosphate inoculation buffer with 500mg of silicon carbide powder (600 grit). The mixture is applied to leaves using cotton swabs or cheesecloth, with sufficient pressure to create minor abrasions without severe tissue damage [78].
Validation of Silencing Efficiency
Effective gene silencing must be confirmed through multiple methods:
- RT-qPCR: Quantify transcript reduction compared to empty vector controls
- Phenotypic Markers: Include known visual markers like Phytoene Desaturase (PDS) which causes photobleaching
- Western Blotting: Confirm reduction at protein level when antibodies are available
- Sequencing: Verify siRNA production from target region [77] [78]
Control groups should always include empty vector controls and non-inoculated plants to distinguish virus symptoms from silencing phenotypes.
Transgenic Complementation Protocol
Vector Design and Transformation
Complementation vectors should contain:
- Full-length genomic sequence including native promoter and regulatory elements
- Selection marker (antibiotic/herbicide resistance)
- Optionally, reporter genes (GFP, GUS) for transformation tracking
For NBS-LRR genes with complex genomic structures, Bacterial Artificial Chromosomes (BACs) may be required to capture large genomic regions with native regulatory sequences.
Plant Transformation Methods
- Agrobacterium-mediated transformation: Most common for dicots
- Biolistic particle delivery: Useful for monocots
- Protoplast transformation: For rapid testing in cell culture
Molecular Characterization of Transformants
- PCR Genotyping: Confirm transgene integration
- Southern Blotting: Determine copy number and integration pattern
- RT-qPCR/Western: Verify expression at transcript/protein level
- Segregation Analysis: Confirm Mendelian inheritance in T2 generation
Application in NBS Gene Evolution Research
Studying Gene Duplication Events
NBS-LRR genes represent one of the largest and most dynamic gene families in plants, characterized by extensive duplication and diversification. VIGS and complementation are invaluable for functional analysis of duplicated genes.
Table 1: VIGS Applications in NBS Gene Functional Analysis
| Research Objective | Experimental Approach | Key Findings | Reference |
|---|---|---|---|
| Role of duplicated NBS genes in disease resistance | Silencing of individual gene copies in resistant cotton | Identified specific NBS genes (OG2) essential for virus resistance | [20] |
| Functional conservation after duplication | VIGS of orthologous NBS genes across species | Duplicated genes maintain pathogen specificity despite sequence divergence | [79] |
| Neofunctionalization of duplicated NBS genes | Silencing recent duplicates in grass pea | Subfunctionalization in stress responses; some copies gain new functions | [79] |
| Expression plasticity of tandem duplicates | Tissue-specific VIGS in different organs | Divergent expression patterns among duplicated genes in roots vs leaves | [78] |
Gene duplication generates genetic novelty through several mechanisms:
- Neofunctionalization: One duplicate acquires new function
- Subfunctionalization: Partitioning of original functions between duplicates
- Conservation: Maintenance of identical functions for dosage effects
VIGS enables functional dissection of these evolutionary trajectories by allowing targeted silencing of individual paralogs. For example, in cotton, VIGS identified specific NBS genes within expanded clusters that confer resistance to Cotton Leaf Curl Disease [20].
Elucidating Evolutionary Patterns
The NBS gene family exhibits distinctive evolutionary patterns driven by duplication mechanisms:
Whole Genome Duplication (WGD):
- Generates large numbers of NBS paralogs
- Genes typically under strong purifying selection (low Ka/Ks ratios)
- Often maintained as core conserved subgroups [11]
Tandem Duplication:
- Creates copy number variation between genotypes
- Associated with relaxed selection or positive selection
- Generates highly variable "adaptive" subgroups [11] [14]
Transgenic complementation tests functional equivalence between orthologs, revealing evolutionary conservation. For instance, complementation with heterologous NBS genes can determine whether sequence divergence corresponds to functional divergence.
Research Reagent Solutions
Table 2: Essential Research Reagents for VIGS and Complementation Studies
| Reagent Category | Specific Examples | Application Notes | References |
|---|---|---|---|
| Viral Vectors | TRV, FoMV, BSMV, TMV | TRV: broad host range; FoMV: optimized for monocots | [77] [78] |
| Agrobacterium Strains | GV3101, LBA4404 | GV3101: superior for Nicotiana benthamiana infiltration | [77] [78] |
| Visual Marker Genes | PDS, ChlI, ChlD | PDS: photobleaching; ChlI/ChlD: chlorophyll deficiency | [77] [78] |
| Cloning Systems | Gateway, LIC, Restriction-based | Gateway: high-throughput; LIC: sequence-independent | [77] |
| Plant Transformation | Binary vectors, reporter tags | pTRV2 (TRV system), pFoMV (FoMV system) | [77] [78] |
| Selection Agents | Kanamycin, Hygromycin | Antibiotic resistance markers for transgenic selection | [78] [80] |
Case Studies in Gene Functional Validation
VIGS Validation of NBS Gene in Cotton
A comprehensive study demonstrated the functional validation of a cotton NBS gene responsible for resistance to Cotton Leaf Curl Disease (CLCuD). Researchers identified NBS genes through comparative genomics between resistant (Mac7) and susceptible (Coker 312) cotton accessions. Virus-induced silencing of a specific NBS gene (GaNBS from orthogroup OG2) in resistant plants resulted in increased viral titers and susceptibility symptoms, confirming its essential role in pathogen defense [20].
SMV Resistance Gene in Soybean
In soybean, researchers identified Glyma02g13380 as a candidate gene conferring resistance to Soybean Mosaic Virus strains SC4 and SC20. VIGS-mediated silencing of this gene in resistant cultivar Kefeng-1 compromised immunity, while transgenic complementation will be needed to definitively confirm gene identity [80].
Salt Stress Response Gene in Cotton
A recent investigation combined transcriptomics with VIGS to identify GhSAP6 as a negative regulator of salt tolerance in upland cotton. Silencing of GhSAP6 enhanced salt tolerance, while overexpression studies confirmed its negative regulatory function. This study exemplifies how VIGS can rapidly identify genes for potential crop improvement [81].
Technical Considerations and Limitations
VIGS Constraints and Optimization
While powerful, VIGS presents several technical challenges:
- Transient Efficacy: Silencing is often transient, lasting 3-8 weeks depending on host-virus combination
- Off-Target Effects: Sequence similarity may cause unintended silencing of non-target genes
- Host Range Limitations: Not all species are amenable to existing viral vectors
- Viral Symptom Interference: Pathogenicity may confound phenotypic analysis [76] [77]
Optimization strategies include:
- Temperature control (21-25°C) to balance viral spread and plant health
- Plant developmental stage selection (younger plants generally more susceptible)
- Vector modification to reduce pathogenicity while maintaining silencing efficiency
- Multiple target fragments to confirm phenotype specificity [77] [78]
Complementation Challenges
Transgenic complementation faces different hurdles:
- Position Effects: Random integration may alter expression patterns
- Gene Silencing: Transgenes may be subject to epigenetic silencing
- Cumbersome Process: Time-consuming, especially for perennial species
- Regulatory Constraints: GMO regulations may limit application
VIGS and transgenic complementation provide complementary approaches for functional validation of genes, particularly in rapidly evolving families like NBS-LRR genes. The integration of these methods with emerging technologies promises to accelerate gene functional characterization:
- CRISPR/VIGS Integration: Using CRISPR for stable knockout with VIGS for rapid screening
- Multiplexed VIGS: Silencing multiple gene family members simultaneously
- Single-Cell Transcriptomics: Resolving expression patterns of duplicated genes
- VIGS-Induced Epigenetic Editing: Leveraging heritable silencing for trait development [76]
For researchers investigating gene duplication events, the combined application of VIGS and complementation offers a powerful toolkit to dissect functional evolution, identify key residues determining specificity, and ultimately engineer improved disease resistance in crop species.
The NBS-LRR gene family constitutes one of the largest classes of disease resistance (R) genes in plants, playing a critical role in detecting pathogens and initiating robust immune responses [82] [5]. These genes encode proteins characterized by a central nucleotide-binding site (NBS) domain and a C-terminal leucine-rich repeat (LRR) domain, with the N-terminal region typically featuring either a Toll/interleukin-1 receptor (TIR) or coiled-coil (CC) domain, classifying them as TNL or CNL types, respectively [59]. The LRR domain facilitates protein-protein interactions and pathogen recognition, while the NBS domain binds nucleotides, providing energy for downstream signaling cascades that often culminate in the hypersensitive response—a programmed cell death at infection sites that restricts pathogen spread [82] [59].
This case study explores the functional characterization of a specific NBS-LRR gene within the context of evolutionary arms races between plants and pathogens. Such conflicts drive relentless cycles of adaptation, where pathogens evolve effectors to suppress plant immunity, and plants counter with diversified recognition capacities [14]. A key mechanism fueling this diversification in NBS-LRR genes is gene duplication, which creates genetic redundancy that allows new gene copies to explore functional mutations without fitness costs [14]. Genomic regions prone to duplication, often associated with specific repeats, can therefore provide an evolutionary advantage by serving as hotbeds for generating novel resistance specificities [14].
We focus on Fusarium wilt disease in tung trees (Vernicia species), caused by the soil-borne fungus Fusarium oxysporum. This disease poses a severe threat to cultivation, particularly to the high-quality oil-producing Vernicia fordii, which is susceptible, while its counterpart, Vernicia montana, exhibits robust resistance [82]. Through a comparative genomics approach, researchers have identified a candidate NBS-LRR gene in V. montana implicated in this resistance, providing a model for studying the molecular basis of disease resistance and its evolution.
Background and Genomic Context
The NBS-LRR Gene Family in Vernicia Species
A systematic genome-wide analysis of two tung tree genomes, V. fordii (susceptible) and V. montana (resistant), identified 239 NBS-LRR genes—90 in V. fordii and 149 in V. montana [82]. The composition of these genes reveals significant structural differences between the two species, as detailed in Table 1.
Table 1: Distribution of NBS-LRR Genes in Vernicia Genomes
| Species | Total NBS-LRR Genes | CC-NBS-LRR | TIR-NBS-LRR | NBS-LRR | CC-NBS | NBS | Other Types |
|---|---|---|---|---|---|---|---|
| V. fordii (Susceptible) | 90 | 12 | 0 | 12 | 37 | 29 | 0 |
| V. montana (Resistant) | 149 | 9 | 3 | 12 | 87 | 29 | 9 |
Notably, no TIR-domain-containing NBS-LRRs (TNLs) were found in the susceptible V. fordii, whereas V. montana possesses 12 genes with TIR domains, including three full-length TNLs [82]. Furthermore, V. montana possesses unique LRR domains (LRR1 and LRR4) that are absent in V. fordii, suggesting that gene loss events in the susceptible species may have compromised its immune repertoire [82].
Evolutionary Dynamics of NBS-LRR Genes
The expansion and contraction of the NBS-LRR family are dynamic evolutionary processes influenced by selective pressures from pathogens. Studies across diverse plant families reveal distinct evolutionary patterns:
- "Consistent Expansion": Observed in potato and some legumes, where the gene family has steadily grown [5].
- "Expansion and Contraction": Seen in tomato and yellowhorn, featuring an initial increase followed by a decrease in gene numbers [5].
- "Sharp Expansion to Abrupt Shrinking": A pattern shared by apple and pear in the Rosaceae family [5].
These patterns are driven by mechanisms such as tandem gene duplication and segmental duplication, often facilitated by duplication-prone genomic regions rich in specific repeats [14]. Lineages where arms-race genes are physically associated with these duplication-inducing sequences enjoy a selective advantage, leading to a measurable statistical association between the two over evolutionary time [14]. This framework of birth-and-death evolution under positive selection provides the context for the identification of specific resistance genes.
Identification and Characterization of the Candidate Gene
Orthologous Gene Pair with Divergent Expression
Comparative genomic analysis between V. fordii and V. montana identified 43 orthologous NBS-LRR pairs [82]. Among these, the orthologous pair Vf11G0978-Vm019719 emerged as a prime candidate. This pair exhibits strikingly distinct expression patterns following Fusarium wilt infection: the V. fordii allele (Vf11G0978) shows downregulated expression, whereas its V. montana ortholog (Vm019719) is significantly upregulated [82]. This differential response suggested that Vm019719 could be a key mediator of resistance in V. montana.
Promoter Analysis Reveals a cis-Regulatory Mutation
Further investigation into the regulatory regions of these alleles uncovered a critical mutation. The promoter of the resistant V. montana allele (Vm019719) contains a functional W-box element, which is a known binding site for WRKY transcription factors [82]. This W-box is activated by VmWRKY64, which is implicated in the defense response. In contrast, the susceptible V. fordii allele (Vf11G0978) possesses a deletion in this W-box element [82]. This loss of a key regulatory sequence in the susceptible genotype explains its inability to activate the defense gene effectively, leading to an ineffective response against the pathogen.
Experimental Validation of Gene Function
Virus-Induced Gene Silencing (VIGS) Protocol
Virus-Induced Gene Silencing (VIGS) was employed as the primary functional validation tool to confirm the role of Vm019719 in Fusarium wilt resistance. VIGS is a powerful reverse-genetics technique that uses a modified virus to trigger sequence-specific degradation of target mRNA, effectively "knocking down" gene expression [82] [83].
Table 2: Key Research Reagents for VIGS and Functional Analysis
| Reagent / Tool | Function / Purpose | Example / Source |
|---|---|---|
| HMMER Software | Identifies candidate NBS-LRR genes using hidden Markov models against conserved domains (e.g., PF00931). | [82] [5] |
| VIGS Vector System | Delivers a fragment of the target gene into plant tissues to induce post-transcriptional gene silencing. | TRV-based vectors are commonly used. |
| Reference Genome | Provides the genomic context for mapping, gene annotation, and synteny analysis. | V. montana and V. fordii sequenced genomes [82]. |
| WRKY64 Expression Construct | Used to demonstrate trans-activation of the candidate gene's promoter. | [82] |
The experimental workflow for the VIGS validation is summarized below:
Diagram 1: VIGS experimental workflow for gene validation.
Detailed Methodology:
- Gene Fragment Cloning: A ~200-300 bp fragment specific to the Vm019719 coding sequence was amplified via PCR and cloned into a VIGS vector (e.g., a Tobacco Rattle Virus (TRV)-based vector like pTRV2) [82] [83].
- Plant Infection: The recombinant vector was introduced into Agrobacterium tumefaciens. The bacterial culture was then infiltrated into the leaves of young V. montana seedlings. Control plants were infiltrated with an empty vector (pTRV2) or a vector containing a non-related gene fragment [82].
- Pathogen Challenge: After giving time for the VIGS system to silence the target gene (typically 2-3 weeks), the silenced plants were inoculated with Fusarium oxysporum spores using a root-dipping method.
- Phenotypic and Molecular Assessment: Disease severity was monitored and scored over time. Key assessments included:
Key Experimental Findings
The VIGS experiment provided direct evidence for the function of Vm019719. V. montana plants in which Vm019719 was silenced showed significantly increased susceptibility to Fusarium wilt compared to control plants [82]. This was characterized by more severe wilting and higher fungal biomass within the tissues. This loss-of-function phenotype confirmed that Vm019719 is necessary for full resistance in V. montana.
Furthermore, transcriptional activation assays confirmed that the Vm019719 promoter was activated by VmWRKY64, and this activation was dependent on the intact W-box element present in the resistant species [82].
Integrated Signaling Pathway and Resistance Mechanism
Based on the experimental data, a model for the resistance mechanism mediated by Vm019719 can be proposed. This model integrates pathogen perception, transcriptional regulation, and defense activation, as illustrated below:
Diagram 2: Proposed resistance mechanism of Vm019719.
In this model:
- Pathogen Recognition: Fusarium oxysporum infection provides an initial signal.
- Transcriptional Activation: The signal leads to the expression or activation of the transcription factor VmWRKY64, which binds to the intact W-box in the promoter of Vm019719 in V. montana, driving its expression.
- Defense Execution: The Vm019719 NBS-LRR protein subsequently perceives a specific pathogen effector (or a host component modified by the effector), leading to its activation. This triggers a downstream defense signaling cascade, resulting in a hypersensitive response and the establishment of resistance.
- Susceptibility in V. fordii: In the susceptible V. fordii, the deletion of the W-box in the promoter of the orthologous gene (Vf11G0978) prevents its effective trans-activation by WRKY factors. Consequently, the defense response is not adequately initiated, leading to disease susceptibility.
Discussion and Implications
A Model of cis-Regulatory Evolution in Disease Resistance
This case study highlights that losing a single cis-regulatory element can be a decisive factor in disease susceptibility. The resistance mechanism in V. montana is not based on a novel protein but on the differential regulation of an orthologous gene [82]. This finding underscores the importance of investigating promoter regions and regulatory variations in breeding programs, in addition to coding sequences.
Broader Context of NBS-LRR Gene Evolution
The duplication and loss of NBS-LRR genes, as observed in the differing repertoires of V. fordii and V. montana, are fundamental to plant-pathogen co-evolution. The association of resistance genes with duplication-prone genomic regions is a conserved evolutionary strategy [14]. These regions, often enriched with tandem repeats, act as diversity generators, allowing the rapid exploration of new mutations and the emergence of novel resistance specificities without compromising existing essential functions [14]. The evolutionary history of the NBS-LRR family across plant lineages—ranging from expansion to contraction—reflects the unique pathogen pressures each lineage has faced [5].
Application in Crop Improvement
The identification and validation of Vm019719 provides a direct resource for marker-assisted breeding. The specific promoter polymorphism, particularly the presence/absence of the critical W-box, can be developed into a molecular marker to screen for resistant genotypes [82]. Furthermore, this knowledge enables alternative strategies:
- Pyramiding R Genes: Combining Vm019719 with other Fusarium wilt resistance genes to create durable resistance.
- Precision Breeding: Using gene editing to restore the function of the susceptible allele or to introduce the functional promoter variant into elite, susceptible cultivars.
This case study demonstrates a comprehensive approach to validating a disease resistance gene, from genome-wide comparative analysis and in silico promoter inspection to functional validation via VIGS. It establishes that the NBS-LRR gene *Vm019719 is a key determinant of Fusarium wilt resistance in Vernicia montana, and its function is critically dependent on a cis-regulatory element lost in the susceptible relative. This work provides both a fundamental understanding of resistance mechanics and a practical tool for crop improvement. It also exemplifies how NBS-LRR genes, through processes of duplication, divergence, and regulatory evolution, serve as a dynamic genomic arsenal in the perpetual arms race between plants and their pathogens.
Gene duplication events are fundamental drivers of evolutionary innovation, providing raw genetic material for the emergence of new functions and specialized traits. In plant genomes, this phenomenon is particularly evident in the evolution of nucleotide-binding site (NBS)-encoding genes, which constitute the largest family of plant disease resistance (R) genes. These genes play a critical role in plant immune responses by recognizing pathogen effectors and activating defense mechanisms [17] [20]. The NBS gene family has undergone significant expansion and contraction across plant lineages through various duplication mechanisms, including whole-genome duplication (WGD) and tandem duplication, resulting in remarkable diversity across species [84] [20].
This technical review examines the evolutionary patterns of NBS genes across three distinct plant genera—Nicotiana, Dendrobium, and cereal species—to elucidate how gene duplication events have shaped their disease resistance capabilities. Nicotiana species, particularly the allotetraploid N. tabacum, provide insights into the consequences of recent polyploidization events [17] [85]. Dendrobium species, valued for their medicinal properties, exemplify lineage-specific gene family dynamics in monocots [29]. Cereal genomes reveal patterns of NBS gene evolution in economically important grass species [84]. Through comparative analysis of these systems, we aim to establish a comprehensive understanding of the relationship between gene duplication and the functional diversification of plant immune genes, offering insights for future disease resistance breeding strategies.
Comparative Genomics of NBS Gene Families
NBS Gene Distribution and Classification
The NBS gene family represents one of the most dynamic and rapidly evolving components of plant genomes. These genes typically encode proteins containing a conserved nucleotide-binding site (NBS) domain and often C-terminal leucine-rich repeats (LRRs), which are responsible for pathogen recognition [17] [82]. Based on their N-terminal domains, NBS-encoding genes are classified into several major subfamilies: TIR-NBS-LRR (TNL), CC-NBS-LRR (CNL), RPW8-NBS-LRR (RNL), and various truncated forms lacking complete domains [84] [20].
Table 1: Comparative Analysis of NBS Gene Families Across Plant Species
| Plant Species | Total NBS Genes | CNL | TNL | RNL | Truncated Forms | Key Evolutionary Features |
|---|---|---|---|---|---|---|
| Nicotiana tabacum | 603 | 150 (CC-NBS)74 (CC-NBS-LRR) | 9 (TIR-NBS)64 (TIR-NBS-LRR) | Not specified | 306 (NBS-only) | Allotetraploid with contributions from parental genomes; WGD-driven expansion [17] |
| N. sylvestris | 344 | 82 (CC-NBS)48 (CC-NBS-LRR) | 5 (TIR-NBS)37 (TIR-NBS-LRR) | Not specified | 172 (NBS-only) | Diploid progenitor species [17] |
| N. tomentosiformis | 279 | 65 (CC-NBS)47 (CC-NBS-LRR) | 7 (TIR-NBS)33 (TIR-NBS-LRR) | Not specified | 127 (NBS-only) | Diploid progenitor species [17] |
| Dendrobium officinale | 74 | 10 | 0 | 0 | 64 | Significant gene degeneration; absence of TNL subclass [29] |
| D. nobile | 169 | 18 | 0 | 0 | 151 | Expanded NBS repertoire compared to D. officinale [29] |
| D. chrysotoxum | 118 | 14 | 0 | 0 | 104 | Intermediate NBS gene count [29] |
| Xanthoceras sorbifolium | 180 | Majority | Minority | Present | Not specified | "First expansion and then contraction" pattern [84] |
| Acer yangbiense | 252 | Majority | Minority | Present | Not specified | "First expansion followed by contraction and further expansion" [84] |
| Dinnocarpus longan | 568 | Majority | Minority | Present | Not specified | Strong recent expansion with highest gene count [84] |
| Vernicia fordii | 90 | 49 (with CC domains) | 0 | 0 | 41 | Susceptible to Fusarium wilt [82] |
| Vernicia montana | 149 | 98 (with CC domains) | 12 (with TIR domains) | 0 | 39 | Resistant to Fusarium wilt; unique TIR-containing genes [82] |
Evolutionary Patterns and Duplication Mechanisms
The expansion and contraction of NBS gene families across plant lineages follow distinct evolutionary patterns influenced by both whole-genome duplication (WGD) and small-scale duplication (SSD) events. In the soapberry family (Sapindaceae), comparative analyses of Xanthoceras sorbifolium, Dinnocarpus longan, and Acer yangbiense reveal three distinct evolutionary patterns: "first expansion and then contraction" in X. sorbifolium, "first expansion followed by contraction and further expansion" in A. yangbiense, and a similar pattern with stronger recent expansion in D. longan [84]. These patterns result from independent gene duplication and loss events following species divergence, with D. longan gaining significantly more genes potentially in response to diverse pathogen pressures [84].
In Nicotiana species, whole-genome duplication has been a major driver of NBS gene family expansion. The allotetraploid N. tabacum contains approximately the combined total of NBS genes from its diploid progenitors (N. sylvestris and N. tomentosiformis), with 76.62% of its NBS genes traceable to these parental genomes [17]. This demonstrates the significant role of polyploidization in generating genetic material for evolutionary innovation.
Table 2: Evolutionary Patterns of NBS Genes Across Plant Lineages
| Evolutionary Pattern | Representative Species | Key Characteristics | Proposed Driving Forces |
|---|---|---|---|
| Allopolyploid Expansion | Nicotiana tabacum | Combines NBS genes from progenitor species; WGD contributes significantly to expansion [17] | Hybridization and genome doubling |
| Lineage-Specific Contraction | Dendrobium officinale | Significant reduction in NBS-LRR genes; absence of TNL subclass; domain degeneration common [29] | Specialized evolutionary trajectory in monocots |
| Progressive Expansion | Dinnocarpus longan | Strong recent expansion with 568 NBS genes; dynamic duplication/loss events [84] | Response to diverse pathogen pressures |
| Differential Expression | Vernicia montana | Retains TIR-containing NBS genes lost in susceptible relative V. fordii [82] | Pathogen-driven selection maintaining specific resistance mechanisms |
| Tandem Array Formation | Cereals and grasses | NBS genes clustered as tandem arrays on chromosomes with few singletons [84] | Local duplications generating gene clusters |
Orchids, represented by Dendrobium species, exhibit significant degeneration of NBS genes, particularly affecting the TNL subclass. No TNL-type genes were identified in six orchid species, consistent with the pattern observed in other monocots [29]. This TIR domain degeneration in monocots is potentially driven by NRG1/SAG101 pathway deficiency [29]. The Dendrobium genus also shows frequent type changes and NB-ARC domain degeneration, contributing to NBS gene diversity [29].
Experimental Approaches for NBS Gene Analysis
Genome-Wide Identification and Classification
The identification and classification of NBS-encoding genes require integrated bioinformatic approaches leveraging conserved protein domains and motif structures.
Protocol 1: HMMER-Based NBS Gene Identification
- Data Acquisition: Obtain genome assemblies and annotated protein sequences from relevant databases (e.g., NCBI, Phytozome, Plaza) [17] [20].
- Domain Search: Perform hidden Markov model (HMM) searches using HMMER v3.1b2 with the PF00931 (NB-ARC) model from the PFAM database [17] [84].
- Domain Confirmation: Verify identified candidates using:
- Classification: Categorize genes based on domain architecture into CNL, TNL, RNL, and truncated forms [20].
- Manual Curation: Remove redundant hits and verify domain completeness through multiple database searches.
Protocol 2: Evolutionary and Phylogenetic Analysis
- Multiple Sequence Alignment: Use MUSCLE v3.8.31 or MAFFT 7.0 with default parameters for protein sequence alignment [17] [20].
- Phylogenetic Reconstruction: Construct trees using MEGA11 or FastTreeMP with maximum likelihood method and 1000 bootstrap replicates [17] [20].
- Orthogroup Delineation: Employ OrthoFinder v2.5.1 with DIAMOND for sequence similarity searches and MCL clustering algorithm for orthogroup identification [20].
- Duplication Analysis:
- Selection Pressure Analysis: Calculate non-synonymous (Ka) and synonymous (Ks) substitution rates using KaKs_Calculator 2.0 with Nei-Gojobori model [17].
Functional Characterization Approaches
Protocol 3: Expression and Functional Analysis
Transcriptomic Profiling:
- Retrieve RNA-seq datasets from NCBI SRA or specialized databases [17]
- Process raw sequencing files with fastq-dump v2.6.3 and quality control with Trimmomatic v0.36 [17]
- Map reads to reference genomes using Hisat2 [17]
- Conduct transcript quantification and differential expression analysis with Cufflinks v2.2.1 and Cuffdiff with FPKM normalization [17]
Virus-Induced Gene Silencing (VIGS):
- Design gene-specific fragments (300-500 bp) for cloning into VIGS vectors [82]
- Transform vectors into Agrobacterium tumefaciens and infiltrate into plant leaves [82]
- Monitor phenotype development and validate silencing efficiency through qRT-PCR [82]
- Challenge silenced plants with pathogens and assess disease symptoms [82]
Promoter Analysis:
The following diagram illustrates the integrated experimental workflow for comprehensive NBS gene analysis:
Figure 1. Comprehensive NBS Gene Analysis Workflow. The diagram outlines the integrated experimental pipeline from genome assembly to breeding applications, highlighting key computational and functional validation steps.
NBS Gene Signaling Pathways and Immune Mechanisms
NBS-LRR genes function as critical components in plant immune signaling pathways, particularly in effector-triggered immunity (ETI). These genes encode receptors that recognize pathogen effectors directly or indirectly, initiating complex signaling cascades that culminate in defense responses [82] [29].
The NBS-LRR proteins can be divided into two major functional classes based on their N-terminal domains: TNLs and CNLs, which may activate somewhat distinct downstream signaling pathways [20]. Recent evidence suggests that RNL-type proteins function downstream of both TNLs and CNLs as common signaling components [84]. Upon pathogen recognition, NBS-LRR proteins undergo conformational changes that activate their signaling potential, leading to downstream responses including mitogen-activated protein kinase (MAPK) activation, hormone signaling modulation, and transcriptional reprogramming [29].
Table 3: Research Reagent Solutions for NBS Gene Studies
| Reagent/Tool | Application | Specifications | Key Features |
|---|---|---|---|
| HMMER v3.1b2 | NBS domain identification | PF00931 (NB-ARC) HMM profile | Identifies distant homologs using hidden Markov models [17] |
| MCScanX | Duplication event analysis | Collinearity detection algorithm | Distinguishes WGD, tandem, and segmental duplications [17] |
| KaKs_Calculator 2.0 | Selection pressure analysis | NG (Nei-Gojobori) model | Calculates Ka/Ks ratios to infer evolutionary forces [17] |
| VIGS Vectors | Functional validation | TRV-based systems | Efficient gene silencing in diverse plant species [82] |
| Hisat2 | Transcriptome mapping | Splice-aware aligner | Accurate alignment of RNA-seq reads to reference genomes [17] |
| OrthoFinder | Evolutionary relationships | MCL clustering algorithm | Discerns orthologs and paralogs across species [20] |
| Dual-Luciferase System | Promoter activity analysis | Firefly/Renilla luciferase | Quantifies transcriptional regulation of NBS genes [86] |
| Yeast One-Hybrid | Protein-DNA interactions | GAL4-based system | Identifies transcription factors regulating NBS genes [82] |
The following diagram illustrates the core signaling pathways involved in NBS-mediated immunity:
Figure 2. NBS-Mediated Immune Signaling Pathways. The diagram illustrates how different NBS receptor types (TNL, CNL) recognize pathogen effectors and converge on RNL helpers to activate downstream defense signaling through MAPK cascades, hormone pathways, and transcriptional reprogramming.
In Dendrobium officinale, NBS-LRR genes participate not only in the ETI system but also in plant hormone signal transduction pathways and the Ras signaling pathway [29]. Transcriptome analysis following salicylic acid (SA) treatment identified 1,677 differentially expressed genes, including six NBS-LRR genes that were significantly upregulated [29]. One gene in particular, Dof020138, showed extensive connectivity to multiple pathways including pathogen recognition, MAPK signaling, plant hormone signal transduction, biosynthetic pathways, and energy metabolism pathways [29].
The functional specialization of NBS genes is evident in comparative studies between resistant and susceptible varieties. In tung trees, the orthologous gene pair Vf11G0978-Vm019719 exhibits distinct expression patterns in Vernicia fordii (susceptible) and V. montana (resistant) [82]. In resistant V. montana, Vm019719 is upregulated and confers resistance to Fusarium wilt when activated by VmWRKY64, while its allelic counterpart in susceptible V. fordii shows an ineffective defense response due to a deletion in the promoter's W-box element [82]. This highlights how regulatory variations in NBS genes can determine disease resistance outcomes.
The cross-species comparative analysis of NBS genes in Nicotiana, Dendrobium, and cereal genomes reveals the profound impact of gene duplication events on the evolution of plant immunity. Whole-genome duplications, as evidenced in Nicotiana tabacum, provide substantial genetic material for functional diversification, while tandem duplications enable rapid adaptation to specific pathogen pressures. The contrasting evolutionary patterns observed—from expansion in Dinnocarpus longan to contraction and degeneration in Dendrobium species—highlight the dynamic nature of plant immune gene repertoires.
The functional characterization of NBS genes across these species has identified key candidates for disease resistance breeding, such as the V. montana Vm019719 gene for Fusarium wilt resistance and D. officinale Dof020138 involved in SA-responsive defense signaling. These findings, coupled with advanced genomic technologies and functional validation tools, provide powerful resources for molecular breeding programs aimed at enhancing crop resilience.
Future research should focus on integrating pan-genome analyses to capture the full diversity of NBS genes within species, elucidating the precise molecular mechanisms of pathogen recognition, and developing precision breeding strategies that stack favorable NBS alleles while maintaining plant fitness. The continued comparative analysis of NBS gene evolution across plant lineages will undoubtedly yield further insights into the complex interplay between gene duplication, functional innovation, and plant-pathogen co-evolution.
Plant survival in naturally pathogenic environments hinges on a sophisticated innate immune system. A critical component of this system is the nucleotide-binding site-leucine-rich repeat (NBS-LRR) gene family, one of the largest plant gene families responsible for encoding major disease resistance (R) proteins [3] [20]. These genes confer resistance by recognizing pathogen effectors and initiating robust defense responses, including a form of programmed cell death at the infection site known as the hypersensitive response [9]. The evolution of NBS-LRR genes is characterized by remarkable dynamism, driven by frequent gene duplication and loss events that enable a rapid adaptation to evolving pathogens [3] [14]. This birth-and-death evolution creates a direct, selectable link between the plant's genotype (the repertoire of NBS-LRR genes) and its phenotype (resistance or susceptibility) [9].
The defense phenotype is orchestrated by complex signaling networks, with the hormone salicylic acid (SA) serving as a master regulator [87] [88]. SA accumulation is essential for establishing both local resistance and systemic acquired resistance (SAR), which provides long-lasting, broad-spectrum protection throughout the plant [88]. The interplay between a plant's rapidly evolving NBS-LRR genotype and the SA-mediated defense signaling network forms the core of understanding how genotype translates into phenotype under pathogen pressure. This guide provides a technical framework for conducting expression analysis to dissect these critical relationships, with a specific focus on the context of NBS gene duplication events.
Core Concepts and Quantitative Landscape
The NBS-LRR Gene Family: Architecture and Evolution
NBS-LRR genes are modular proteins typically consisting of a variable N-terminal domain, a central conserved NBS (NB-ARC) domain, and a C-terminal LRR domain [20]. They are classified into subfamilies based on the N-terminal domain:
- TNLs: Contain a Toll/Interleukin-1 Receptor (TIR) domain.
- CNLs: Contain a Coiled-Coil (CC) domain.
- RNLs: Contain a Resistance to Powdery Mildew 8 (RPW8) domain and often function in signaling downstream of TNLs and CNLs [3] [20].
A genome-wide analysis of 12 Rosaceae species revealed 2,188 NBS-LRR genes, showcasing dramatic variation in copy numbers between species due to independent gene duplication and loss events [3]. This expansion is not random; studies in barley indicate that natural selection favors lineages where these "arms-race genes" are physically associated with duplication-prone genomic regions, facilitating the efficient generation of new resistance specificities [14].
Salicylic Acid: A Master Regulator of Immunity
SA is a phenolic phytohormone synthesized primarily via the isochorismate (IC) pathway in plants like Arabidopsis, with a secondary role for the phenylalanine ammonia-lyase (PAL) pathway [87] [88]. Its role in defense is twofold:
- Signal Perception: SA is perceived by a suite of receptors, including NPR1 (Nonexpressor of Pathogenesis-Related genes 1) and its paralogs NPR3 and NPR4, leading to the expression of defense-related genes [87] [88].
- Growth-Defense Trade-offs: SA levels have a profound impact on plant growth. High SA levels often result in a stunted phenotype, as observed in mutants like acd6 and cpr5, while SA-deficient plants can exhibit increased biomass [87]. This highlights a critical trade-off between investment in defense and growth.
Table 1: Summary of NBS-LRR Genes Across Plant Species
| Species | Total NBS-LRR Genes | TNLs | CNLs/Non-TNLs | RNLs | Key Evolutionary Pattern |
|---|---|---|---|---|---|
| Rosaceae (12 species) | 2,188 (combined) | 26 (ancestral TNLs) | 69 (ancestral CNLs) | 7 (ancestral RNLs) | Dynamic; species-specific (e.g., "expansion and contraction") [3] |
| Fragaria spp. (6 species) | 1,134 (combined) | 38 gene families | 146 gene families | Not specified | Lineage-specific duplications pre-dating species divergence [9] |
| Blueberry | 97 | 11 | 86 | Included in Non-TNL | >22% of genes present in clusters [35] |
| Barley | Not specified | Not specified | Not specified | Not specified | Association with duplication-prone genomic regions [14] |
| Maize (Pan-genome) | Not specified | Not specified | Not specified | Not specified | "Core-adaptive" model with Presence-Absence Variation [11] |
Table 2: Effect of Exogenous Salicylic Acid on Plant Growth in Different Species
| Plant Species | SA Concentration | Effect on Growth | Biological Context |
|---|---|---|---|
| African Violet | 0.01 mM | Increased rosette diameter, leaf & flower bud number | Promotive effect [87] |
| Wheat | 0.05 mM | Stimulated seedling growth and larger ears | Promotive effect [87] |
| Chamomilla | 0.05 mM | Stimulated leaf (32%) and root (65%) growth | Promotive effect [87] |
| Chamomilla | 0.25 mM | Decreased leaf (40%) and root (43%) growth | Inhibitory effect [87] |
| Arabidopsis | < 0.05 mM | Promoted adventitious root formation | Promotive effect [87] |
| Arabidopsis | > 0.05 mM | Inhibited all root growth processes | Inhibitory effect [87] |
| Tobacco | 0.1 mM | Reduced shoot growth and leaf epidermal cell size | Inhibitory effect [87] |
Technical Guide: Expression Analysis Workflow
This section outlines a comprehensive experimental workflow for linking NBS-LRR genotypes to defense phenotypes through expression analysis under pathogen and SA treatment.
Experimental Design and Preparation
A. Defining the Genotypic Contrast The first step is to select plant materials with contrasting NBS-LRR genotypes and/or resistance phenotypes. Effective comparisons include:
- Resistant vs. Susceptible Cultivars: For example, in cotton, compare the tolerant G. hirsutum accession 'Mac7' with the susceptible 'Coker 312'. Mac7 possesses numerous unique genetic variants in its NBS genes, which are candidate loci for the resistant phenotype [20].
- Mutant Lines: Utilize SA biosynthesis mutants (e.g., sid2, NahG transgenic lines) or signaling mutants (e.g., npr1) alongside wild-type controls to disentangle the contribution of SA [87] [88].
B. Treatment Application
- Pathogen Inoculation: Use a standardized protocol for the pathogen of interest (e.g., bacterial spray infiltration, fungal spore application). For viral pathogens like the cotton leaf curl virus, use whitefly-mediated transmission or agro-infiltration [20].
- Hormone Treatment: Prepare a fresh SA solution. A common working concentration is 0.5-1.0 mM, but dose-response experiments are recommended as effects are concentration-dependent (see Table 2). Include a mock treatment (e.g., water with a small amount of solvent like ethanol) as a control [87].
- Time-Course Sampling: Collect tissue samples at multiple time points post-treatment (e.g., 0, 6, 12, 24, 48, and 72 hours) to capture the dynamics of the immune response. Flash-freeze samples in liquid nitrogen and store at -80°C.
Methodologies for Gene Expression Analysis
A. RNA Extraction and Sequencing
- Protocol: Extract total RNA using a commercial kit (e.g., TRIzol-based methods or silica-membrane columns) with an on-column DNase I digestion step to remove genomic DNA contamination. Assess RNA integrity (RNA Integrity Number, RIN > 8.0) using an Agilent Bioanalyzer.
- Library Prep and Sequencing: Prepare stranded mRNA-seq libraries from 1 µg of high-quality total RNA. Sequence on an Illumina platform to generate a minimum of 20 million paired-end (e.g., 150 bp) reads per sample.
B. Transcriptomic Data Analysis
- Read Processing and Alignment: Quality-trim raw reads using Trimmomatic. Align the cleaned reads to the respective reference genome using splice-aware aligners like STAR or HISAT2.
- Quantification and Differential Expression: Generate read counts for each gene feature using featureCounts. Perform differential expression analysis with tools like DESeq2 or edgeR in R. A typical design formula would be:
~ genotype + treatment + genotype:treatment. The interaction term is crucial for identifying genes that respond differently to treatment between genotypes. - Focus on NBS-LRR Genes: Extract expression values (e.g., FPKM or TPM) for annotated NBS-LRR genes from your dataset. In blueberry and other species, these genes can be identified by the presence of the NB-ARC domain (PF00931) [35]. Orthogroup analysis, as performed across land plants, can help identify conserved and lineage-specific NBS-LRR genes for focused study [20].
C. Functional Validation via Gene Silencing
- Virus-Induced Gene Silencing (VIGS): To confirm the functional role of a candidate NBS-LRR gene, use VIGS. Clone a 300-500 bp fragment of the target gene into a VIGS vector (e.g., TRV-based pYL156). Agro-infiltrate this construct into young seedlings. After 2-3 weeks, challenge the silenced plants with the pathogen and quantify disease symptoms and pathogen titer. Silencing of GaNBS (OG2) in resistant cotton demonstrated its role in reducing virus titer [20].
Diagram 1: Experimental workflow for linking genotype to phenotype.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Expression Analysis
| Reagent/Resource | Function/Application | Technical Notes |
|---|---|---|
| SA (Salicylic Acid) | Chemical inducer of defense responses and SAR. | Prepare a stock solution in ethanol or NaOH; use appropriate mock controls. Concentration is critical (see Table 2) [87]. |
| NahG Transgenic Line | A control genotype that degrades SA, abolishing SA-mediated signaling. | Crucial for determining SA-dependent vs. SA-independent responses [87] [88]. |
| VIGS (VIGS Vector) | Functional validation of candidate NBS-LRR genes via transient silencing. | TRV-based vectors (e.g., pYL156) are widely used. Requires a 300-500 bp gene fragment [20]. |
| Reference Genome & Annotation | Essential for RNA-seq read alignment and gene quantification. | Must include well-annotated NBS-LRR genes, identified via NB-ARC (PF00931) HMM profiles [3] [20] [35]. |
| Orthogroup (OG) Classifications | Evolutionary framework for comparing NBS-LRR genes across species. | Allows researchers to focus on conserved (e.g., OG2, OG6, OG15) or lineage-specific gene clusters [20]. |
Data Interpretation and Integration with Gene Duplication
Analyzing Expression Data in an Evolutionary Context
When interpreting your expression data, frame the results within the evolutionary history of NBS-LRR genes.
- Core vs. Adaptive Clades: As observed in the maize pan-genome, "core" NBS genes (e.g., ZmNBS31) are often constitutively expressed and may play fundamental roles in basal immunity. In contrast, genes from "adaptive" clades, which exhibit presence-absence variation, are more likely to show pathogen- or genotype-specific induction [11].
- Expression of Tandem Duplicates: Genes arising from recent tandem duplications, often under diversifying selection, may show neofunctionalization or subfunctionalization. This can be reflected in their expression patterns—for instance, one copy may retain the original expression profile while another acquires a new induction pattern [9] [14]. In strawberries, TNLs were found to be under stronger diversifying selection than non-TNLs, which could correlate with more dynamic expression patterns [9].
- Orthogroup-Centric Analysis: Rather than analyzing individual genes, consider the expression profile of entire orthogroups. In cotton, entire orthogroups (OG2, OG6, OG15) were upregulated in response to cotton leaf curl disease, highlighting conserved functional roles across species [20].
Placing SA Signaling in the Pathway
The expression of NBS-LRR genes and the activation of SA signaling are interconnected. The diagram below integrates the key components of this network, which should be considered when interpreting transcriptomic data.
Diagram 2: NBS-LRR and SA signaling pathway integration.
Concluding Remarks
Linking the genotype to phenotype in plant-pathogen interactions requires a sophisticated approach that integrates evolutionary genetics, molecular biology, and functional genomics. This guide has outlined a pathway for conducting expression analyses that explicitly connect the dynamic evolutionary history of NBS-LRR genes—shaped by duplication and selection—with the SA-mediated defense phenotype. By employing a comparative genotypic framework, high-resolution transcriptomics, and robust functional validation, researchers can move beyond simple catalogs of gene lists to a mechanistic understanding of how specific genetic architectures, forged by evolution, confer a definable resistance phenotype. This knowledge is fundamental for the future-directed breeding and bioengineering of durable disease resistance in crops.
Conclusion
Gene duplication events, particularly tandem and whole-genome duplication, are fundamental forces driving the expansion and functional diversification of the NBS-LRR gene family. This evolutionary strategy enables plants to rapidly generate genetic novelty essential for recognizing evolving pathogens, a classic arms race. The integration of advanced bioinformatic methodologies with robust experimental validation, such as VIGS, is crucial for moving from genetic prediction to confirmed function. Future research should focus on harnessing this knowledge for marker-assisted breeding and genome editing to develop durable disease-resistant crops. Furthermore, exploring the 'cooperative' relationship between duplication-inducing genomic elements and arms-race genes presents a promising frontier for understanding and engineering plant immunity.
References
- 1. Plant NBS-LRR proteins: adaptable guards - Genome Biology [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2006-7-4-212]
- 2. Plant NBS-LRR proteins in pathogen sensing and host defense [https://pmc.ncbi.nlm.nih.gov/articles/PMC1973153/]
- 3. Genome-wide analysis of NBS-LRR genes in Rosaceae ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9685399/]
- 4. Evolution of Gene Duplication in Plants - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4972278/]
- 5. Genome-wide analysis of NBS-LRR genes in Rosaceae ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2022.1052191/full]
- 6. Genome-Wide Characterization and Expression Pattern ... [https://www.mdpi.com/1422-0067/26/18/9063]
- 7. Genome-Wide Identification of NBS-LRR Family in Three ... [https://www.mdpi.com/2073-4425/16/6/680]
- 8. Genome-wide identification, characterization, and ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111922009374]
- 9. Lineage-specific duplications of NBS-LRR genes occurring ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4521-4]
- 10. Contrasting patterns of evolution following whole genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3246211/]
- 11. Evolutionary Conservation and Pre-Activated Immunity of ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1656786/full]
- 12. Short tandem gene duplications as potential agents ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607827/]
- 13. The impact of tandem duplication on gene evolution in ... [https://www.sciencedirect.com/science/article/pii/S2095311921636985]
- 14. Do duplication-inducing elements 'cooperate' with genes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12573847/]
- 15. Impact of whole-genome duplications on structural variant ... [https://www.nature.com/articles/s41467-024-49679-y]
- 16. DTDHM: detection of tandem duplications based on hybrid ... [https://peerj.com/articles/17748/]
- 17. Genome-Wide Identification of NBS-LRR Family in Three ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12192410/]
- 18. Genome-wide identification and characterization of NBS ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-020-02803-8]
- 19. Genome-wide identification of GhEDS1 gene family members ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-07243-w]
- 20. Comparative analysis, diversification, and functional ... [https://www.nature.com/articles/s41598-024-62876-5]
- 21. Phylogenetic, Structural, and Evolutionary Insights into ... [https://www.mdpi.com/1422-0067/26/5/1828]
- 22. A compendium of genome-wide sequence reads from NBS ... [https://www.nature.com/articles/s41598-020-67848-z]
- 23. Rethinking the role of gene duplication on short ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12281259/]
- 24. Exploring Cloned Disease Resistance Gene Homologues and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12385795/]
- 25. Do duplication-inducing elements 'cooperate' with genes in ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-07328-6]
- 26. Identification and Characterization of NBS Resistance ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.758559/full]
- 27. Deciphering Plant NLR Genomic Evolution: Synteny- ... [https://pubmed.ncbi.nlm.nih.gov/39835721/]
- 28. Evolutionary balance between LRR domain loss and young ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-6212-1]
- 29. Evolution patterns of NBS genes in the genus Dendrobium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9661794/]
- 30. Characterizing lineage-specific evolution and the processes ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/s12862-020-1585-y]
- 31. Genome-Wide Identification and Functional Evolution of NLR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562843/]
- 32. HMMerThread: Detecting Remote, Functional Conserved ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3053371/]
- 33. Gene Duplication and the Structure of Eukaryotic Genomes [https://pmc.ncbi.nlm.nih.gov/articles/PMC311031/]
- 34. An Overview of Duplicated Gene Detection Methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC7565063/]
- 35. Genome-scale examination of NBS-encoding genes in ... [https://www.nature.com/articles/s41598-018-21738-7]
- 36. MCScanX: Multiple Collinearity Scan toolkit X version. The ... [https://github.com/wyp1125/MCScanX]
- 37. MCScanX: a toolkit for detection and evolutionary analysis of ... [https://pubmed.ncbi.nlm.nih.gov/22217600/]
- 38. MCScanX: a toolkit for detection and evolutionary analysis ... [https://www.semanticscholar.org/paper/MCScanX%3A-a-toolkit-for-detection-and-evolutionary-Wang-Tang/93a83472e1389fac3c6d794837308002f6e1112d]
- 39. Protocol for HSDFinder: Identifying, annotating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8243146/]
- 40. HSDFinder: A BLAST-Based Strategy for Identifying Highly ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2021.803176/full]
- 41. Next-generation transcriptome assembly and analysis [https://www.sciencedirect.com/science/article/abs/pii/S1046202318303803]
- 42. Transcriptome Analysis Indicates Considerable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4256766/]
- 43. Genome-wide Identification and Evolutionary Analysis of ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.771814/full]
- 44. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 45. RNA sequencing reveals novel lncRNA modulators in ... [https://www.nature.com/articles/s41598-025-24202-5]
- 46. Identification and comparative analysis of long non-coding ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08539-z]
- 47. Genome-wide identification, characterization, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6195493/]
- 48. RNA-Seq Comprehensive Analysis Reveals the Long ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9403536/]
- 49. Lesson5: Visualizing clusters with heatmap and dendrogram [https://bioinformatics.ccr.cancer.gov/docs/data-visualization-with-r/Lesson5_intro_to_ggplot/]
- 50. Extensive Error in the Number of Genes Inferred from Draft ... [https://journals.plos.org/ploscompbiol/article%3Fid%3D10.1371/journal.pcbi.1003998]
- 51. Mitigating assembly and switch errors in phased genomes ... [https://www.nature.com/articles/s41437-025-00803-8]
- 52. Genome-Wide Identification and Evolutionary Analysis of NBS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7224235/]
- 53. Systematic identification of pseudogenes through ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1636364/]
- 54. A computational approach for identifying pseudogenes in the ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2006-7-s1-s13]
- 55. the effect of structural gene annotation on orthology inference [https://pmc.ncbi.nlm.nih.gov/articles/PMC12263111/]
- 56. Tips for improving genome annotation quality [https://www.maxapress.com/article/id/67e36fa6fa6c5866a7a3e810]
- 57. Genome-Wide Identification and Expression Analysis of NBS ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0107987]
- 58. Durable panicle blast-resistance gene Pb1 encodes an ... [https://pubmed.ncbi.nlm.nih.gov/20807214/]
- 59. Genome-wide identification and characterization of NBS ... [https://www.nature.com/articles/s41598-025-03507-5]
- 60. Genome-Wide Characterization and Expression Pattern ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12469828/]
- 61. Evolution patterns of NBS genes in the genus Dendrobium ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-022-03904-2]
- 62. Divergence and Conservative Evolution of XTNX Genes in ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2017.01844/full]
- 63. PRGminer: harnessing deep learning for the prediction of ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1411525/full]
- 64. Comparative analysis of the NLR gene family in the genomes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507765/]
- 65. Different patterns of gene structure divergence following gene ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-14-652]
- 66. Comparative Genomics Reveals Gene Duplication and ... [https://www.mdpi.com/2311-7524/11/2/209]
- 67. The molecular evolutionary basis of species formation ... [https://www.sciencedirect.com/science/article/pii/S0168952525001672]
- 68. Cluster analysis and data visualization of large-scale gene ... [https://pubmed.ncbi.nlm.nih.gov/9697170/]
- 69. Comprehensive Guide to Cluster Analysis [https://www.displayr.com/understanding-cluster-analysis-a-comprehensive-guide/]
- 70. Semantic design of functional de novo genes from a ... [https://www.nature.com/articles/s41586-025-09749-7]
- 71. RepeatsDB in 2025: expanding annotations of structured ... [https://pubmed.ncbi.nlm.nih.gov/39475178/]
- 72. Reporting Short Tandem Repeats in VarSeq [https://www.goldenhelix.com/blog/reporting-short-tandem-repeats-in-varseq/]
- 73. PROST: quantitative identification of spatially variable ... [https://www.nature.com/articles/s41467-024-44835-w]
- 74. Lineage-specific duplications of NBS-LRR genes occurring ... [https://www.lifescience.net/publications/1553659/lineage-specific-duplications-of-nbs-lrr-genes-occ/]
- 75. Additional Diagnostic Yield through the Analysis of Short ... [https://www.sciencedirect.com/science/article/abs/pii/S1525157825001722]
- 76. Virus-Induced Gene Silencing (VIGS): A Powerful Tool for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10057534/]
- 77. A Methodological Advance of Tobacco Rattle Virus ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.671091/full]
- 78. Foxtail mosaic virus-induced gene silencing (VIGS) in ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-022-00903-0]
- 79. Identification, characterization, and validation of NBS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10318170/]
- 80. Identification and functional validation of a new gene ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2024.1518829/full]
- 81. Comparative transcriptome analysis and functional ... [https://www.sciencedirect.com/science/article/abs/pii/S098194282401074X]
- 82. Functional characterization of NBS-LRR genes reveals an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10898138/]
- 83. Quantitative Trait Locus Mapping and Identification of ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8965654/]
- 84. Distinct Evolutionary Patterns of NBS-Encoding Genes in ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.00737/full]
- 85. Comparative Chloroplast Genomes of Nicotiana Species ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9295722/]
- 86. A chromosome-level Dendrobium moniliforme genome ... [https://www.sciencedirect.com/science/article/pii/S2211383525001236]
- 87. Action of Salicylic Acid on Plant Growth [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.878076/full]
- 88. How does the multifaceted plant hormone salicylic acid ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-017-0364-8]