From Moss to Crop: Evolutionary Trajectories of NLR Immune Genes Across Land Plants
This article provides a comprehensive analysis of the evolution of Nucleotide-binding Leucine-rich Repeat (NLR) genes, the cornerstone of the plant intracellular immune system.
From Moss to Crop: Evolutionary Trajectories of NLR Immune Genes Across Land Plants
Abstract
This article provides a comprehensive analysis of the evolution of Nucleotide-binding Leucine-rich Repeat (NLR) genes, the cornerstone of the plant intracellular immune system. Tracing their trajectory from early land plants like mosses to modern dicots, we explore foundational concepts, including the massive expansion and diversification of NLR repertoires. We detail current methodological approaches for NLR discovery and functional validation, such as pangenomic analysis and high-throughput transformation. The review also addresses key challenges in NLR regulation, including avoiding autoimmunity, and examines comparative genomic studies that reveal the impact of domestication on NLR diversity. Finally, we discuss the translational potential of engineered NLRs for developing durable disease resistance in crops, offering insights for researchers in plant science and related biomedical fields.
The Deep Evolutionary History of Plant NLR Immune Receptors
In the evolutionary history of land plants, the emergence of intracellular immune receptors known as Nucleotide-binding Leucine-rich Repeat Receptors (NLRs) represents a critical adaptation to pathogen pressures. These receptors form a central component of the plant innate immune system, enabling recognition of pathogen effector proteins and activation of robust defense responses [1]. Plants and animals possess NLRs that play pivotal roles in innate immunity; however, comparative genomic analyses indicate that plant and animal NLRs have independently arisen through convergent evolution rather than shared ancestry [1] [2]. The modular architecture of NLR proteins has been evolutionarily conserved across land plants, from bryophytes to dicots, while exhibiting remarkable diversification in sequence and function [3]. This review provides a comprehensive technical analysis of NLR domain architecture, classification into major subfamilies, and evolutionary patterns across plant lineages, with specific methodological guidance for researchers investigating this critical gene family.
Core NLR Domain Architecture
Plant NLR proteins follow a prototypical tripartite domain structure that facilitates their function as molecular switches in immune signaling. The core architecture consists of three conserved domains, each with distinct functional characteristics.
N-terminal Domain
The variable N-terminal domain serves as the primary signaling module and defines the major NLR subclasses. In seed plants, three characteristic N-terminal domains have been identified [3] [4]:
- TIR (Toll/Interleukin-1 Receptor) domain: Found in TNLs, often associated with enzymatic activity
- CC (Coiled-Coil) domain: Found in CNLs, frequently involved in oligomerization
- RPW8 (Resistance to Powdery Mildew 8) domain: Found in RNLs, functioning in signal transduction
This domain is responsible for initiating downstream signaling cascades following receptor activation and exhibits the highest sequence diversity among NLRs, reflecting its adaptation to specific signaling environments [4].
Central Nucleotide-Binding Domain
The central Nucleotide-Binding (NB) domain, also referred to as the NB-ARC domain (Nucleotide-Binding adaptor shared with APAF-1, plant Resistance proteins, and CED-4), belongs to the STAND (Signal Transduction ATPases with Numerous Domains) family of ATPases [4]. This domain functions as a nucleotide-dependent molecular switch with two conserved states:
- ADP-bound state: Maintains the NLR in an inactive conformation
- ATP-bound state: Represents the active signaling conformation
The NB-ARC domain contains several conserved motifs critical for immune function, including the P-loop (involved in nucleotide binding), GLPL, MHD, and Kinase 2 motifs [5]. Nucleotide-dependent conformational changes regulate the transition between inactive and active states, controlling NLR signaling capacity [4].
C-terminal Leucine-Rich Repeat (LRR) Domain
The C-terminal Leucine-Rich Repeat (LRR) domain serves dual functions in NLR proteins:
- Autoinhibition: In the absence of pathogens, the LRR domain interacts tightly with the NB-ARC domain, maintaining the receptor in an ADP-bound inactive state [4]
- Effector Recognition: The LRR domain provides a versatile structural platform for direct or indirect recognition of pathogen effectors, with hypervariable regions determining recognition specificity [2]
The modular architecture of NLRs allows for functional specialization while maintaining core signaling mechanisms, with intramolecular interactions between domains providing safeguards against spurious activation [2].
Major NLR Subfamilies: CNLs, TNLs, and RNLs
Based on N-terminal domain composition, plant NLRs are classified into three major subfamilies with distinct structural features and signaling mechanisms.
CNL Subfamily (CC-NBS-LRR)
CNLs are characterized by an N-terminal Coiled-Coil (CC) domain and represent one of the most abundant NLR classes across land plants. The CC domain typically consists of a bundle of alpha-helices that facilitate protein-protein interactions. Recent structural studies have revealed that the CC domains of certain CNLs (e.g., Arabidopsis ZAR1) are structurally similar to bacterial pore-forming toxins and form pentameric resistosomes that directly insert into plasma membranes, creating calcium-permeable channels that initiate immune signaling and cell death [4] [2]. CNLs can function as either "sensor" NLRs that directly or indirectly recognize pathogen effectors, or as "helper" NLRs that amplify immune signals [3].
TNL Subfamily (TIR-NBS-LRR)
TNLs contain an N-terminal TIR (Toll/Interleukin-1 Receptor) domain that often exhibits enzymatic activity. The TIR domain can generate specific signaling molecules and initiate immune signaling cascades. Structural analyses of TNLs (e.g., RPP1 and ROQ1) have demonstrated that they form oligomeric resistosomes upon activation [4]. Unlike CNLs that can directly form plasma membrane channels, TNL signaling typically requires downstream components, particularly EDS1 (Enhanced Disease Susceptibility 1) family proteins, which form heterodimers with helper NLRs to transduce immune signals [3] [4]. TNLs are largely absent from monocot genomes but expanded significantly in dicot lineages [2].
RNL Subfamily (RPW8-NBS-LRR)
RNLs represent a specialized class of NLRs characterized by an N-terminal RPW8 domain that resembles the N-terminal domains of mammalian MLKL and fungal HELL domains, all of which can form membrane pores and induce cell death [2]. Unlike CNLs and TNLs that primarily function as "sensor" NLRs, RNLs typically act as "helper" NLRs that transduce immune signals downstream of sensor NLRs [3]. RNLs are generally less numerous in plant genomes compared to CNLs and TNLs, often appearing in single-digit counts [5]. They play a crucial role in mediating signal transduction from multiple sensor NLRs, forming a signaling hub in plant immune networks [3].
Table 1: Comparative Features of Major Plant NLR Subfamilies
| Feature | CNLs | TNLs | RNLs |
|---|---|---|---|
| N-terminal domain | Coiled-Coil (CC) | TIR (Toll/Interleukin-1 Receptor) | RPW8 |
| Primary function | Sensor or helper | Sensor | Helper |
| Signaling mechanism | Resistosome formation, membrane channel | Resistosome formation, EDS1-dependent | Pore formation, signal transduction |
| Representative members | ZAR1, RPS2 | RPP1, ROQ1 | NRG1, ADR1 |
| Presence in monocots | Abundant | Largely absent | Present |
| Presence in dicots | Abundant | Abundant | Present |
Evolutionary History and Genomic Distribution
The evolutionary trajectory of NLR genes reveals a complex history of expansion, contraction, and diversification correlated with ecological adaptation and plant colonization of land.
Origin and Diversification
Plant NLRs originated in green algae, where only a few NLR genes have been detected, and massively expanded after plants colonized land approximately one billion years ago [3] [2]. This expansion is evident in the genomic record:
- Charophyte algae: Fewer than a dozen NLRs [2]
- Bryophytes (e.g., Physcomitrella patens): Approximately 25 NLRs [1]
- Lycophytes (e.g., Selaginella moellendorffii): As few as 2 NLRs [1]
- Flowering plants: Dozens to hundreds (e.g., 151 in Arabidopsis thaliana, 459 in wine grape) [1]
This pattern suggests that NLR diversification accelerated with terrestrial colonization, possibly in response to increased pathogen pressure in new ecological niches [3].
Lineage-Specific Expansion and Contraction
NLR gene families exhibit remarkable variability in size across plant species, independent of genome size or phylogenetic position [1]. This variability reflects species-specific evolutionary dynamics:
Table 2: NLR Repertoire Size Variation Across Plant Species
| Species | Common Name | Total NLRs | TNLs | CNLs | Other NLRs |
|---|---|---|---|---|---|
| Arabidopsis thaliana | Thale cress | 151 | 94 | 55 | 0 [1] |
| Oryza sativa | Rice | 458 | 0 | 274 | 182 [1] |
| Physcomitrella patens | Moss | 25 | 8 | 9 | 8 [1] |
| Vitis vinifera | Wine grape | 459 | 97 | 215 | 147 [1] |
| Zea mays | Maize | 95 | 0 | 71 | 23 [1] |
| Glycine max | Soybean | 319 | 116 | 20 | NA [6] |
Notably, TNLs are predominantly found in dicots and are largely absent from most monocot genomes, suggesting lineage-specific loss or expansion [2]. Recent studies on asparagus species demonstrate how NLR repertoires can contract during domestication, with wild species (A. setaceus) harboring 63 NLRs compared to just 27 in cultivated garden asparagus (A. officinalis), potentially explaining increased disease susceptibility in domesticated lines [5].
Genomic Organization and Adaptive Evolution
NLR genes are frequently organized in clusters throughout plant genomes and display rapid birth-death evolution, with frequent gene duplications, rearrangements, and losses [2]. This dynamic genomic organization facilitates the generation of novel recognition specificities through domain shuffling, ectopic recombination, and diversifying selection. The high diversity of NLR genes, both among family members within a single genome and between individuals of the same species, reflects continuous adaptation to evolving pathogen populations [2]. This rapid evolution is driven by ecological adaptation to local pathogen pressures, resulting in species-specific NLR repertoires optimized for particular environmental conditions [3].
Experimental Methodologies for NLR Identification and Characterization
Comprehensive analysis of NLR genes requires integrated genomic, transcriptomic, and functional approaches. Below are detailed protocols for systematic NLR identification and characterization.
Genome-Wide Identification of NLR Genes
Dual-Approach Sequence Identification:
- HMMER-based searches: Perform Hidden Markov Model (HMM) searches using the conserved NB-ARC domain (Pfam: PF00931) as query against target proteomes [5]
- BLAST-based identification: Conduct local BLASTp analyses (BLAST+ v2.0 or newer) against reference NLR protein sequences from well-characterized species (e.g., Arabidopsis thaliana, Oryza sativa), applying a stringent E-value cutoff of 1e-10 [5]
- Candidate consolidation: Combine sequences identified through both methods and extract using bioinformatics tools such as TBtools [5]
Domain Validation and Classification:
- Domain architecture analysis: Validate candidate sequences using InterProScan and NCBI's Batch CD-Search, retaining sequences containing the NB-ARC domain (E-value ≤ 1e-5) as bona fide NLR genes [5]
- Subfamily classification: Query Pfam and PRGdb 4.0 databases to classify NLRs into subfamilies based on complete domain architecture [5]
- Manual curation: Visually inspect domain organization to identify atypical or truncated NLR variants (e.g., NL, CN, RN, TN) that may lack specific domains but retain functional classification [5]
Motif and Conserved Domain Analysis
Conserved Motif Identification:
- MEME Suite analysis: Use MEME suite (meme-suite.org/meme/tools/meme) with motif number set to 10 while maintaining default parameters to identify conserved motifs within NBS domains [5]
- Motif visualization: Generate visual representations of motif distributions using TBtools or similar bioinformatics platforms
- Gene structure analysis: Annotate gene structures (exons, introns, UTRs) through GSDS 2.0 (Gene Structure Display Server) to correlate motif position with gene architecture [5]
Promoter cis-Element Analysis:
- Promoter sequence extraction: Isolate 2000 bp genomic sequences upstream of initial ATG codons for all identified NLR genes [5]
- Regulatory element identification: Process sequences through PlantCARE database to identify cis-acting regulatory elements responsive to defense signals and phytohormones [5]
- Visualization: Generate distribution maps and heat maps of cis-elements using TBtools to identify regulatory patterns [5]
Phylogenetic and Evolutionary Analysis
Phylogenetic Reconstruction:
- Sequence alignment: Perform multiple sequence alignment of NLR protein sequences using Clustal Omega or MAFFT [5]
- Tree construction: Build phylogenetic trees using maximum likelihood method based on the JTT matrix-based model implemented in MEGA software [5]
- Statistical support: Assess node support with 1000 bootstrap replicates to evaluate tree robustness [5]
Orthology and Synteny Analysis:
- Orthogroup clustering: Use OrthoFinder v2.2.7 or similar tools to cluster orthologous NLR genes across species based on sequence similarity, normalizing BLAST bit scores based on gene length and phylogenetic distance [5]
- Collinearity analysis: Perform interspecies comparisons using "One Step MCScanX" in TBtools to identify syntenic NLR regions [5]
- Evolutionary dynamics: Calculate expansion/contraction rates of NLR gene families using CAFE (Comparative Analysis of Gene Family Evolution) or similar tools
Table 3: Essential Research Reagents and Resources for NLR Studies
| Reagent/Resource | Function/Application | Example Tools/Databases |
|---|---|---|
| Genome Databases | Source of genomic and annotation data for NLR identification | Plant GARDEN, Dryad Digital Repository, Phytozome [5] |
| Domain Databases | Domain identification and classification | Pfam, InterPro, PRGdb 4.0 [5] |
| HMMER Suite | Hidden Markov Model searches for conserved domains | HMMER v3.0+ with PF00931 profile [5] |
| BLAST+ | Sequence similarity searches for NLR identification | BLAST+ v2.0+ with custom NLR databases [5] |
| TBtools | Integrated toolkit for genomic data analysis | Gene extraction, visualization, collinearity analysis [5] |
| MEME Suite | Conserved motif identification and analysis | MEME, FIMO, Tomtom for motif discovery [5] |
| PlantCARE | Identification of cis-acting regulatory elements | Promoter analysis, hormone-responsive elements [5] |
| OrthoFinder | Orthogroup inference and comparative genomics | Orthologous group clustering across species [5] |
| MEGA Software | Phylogenetic analysis and tree construction | Maximum likelihood trees, bootstrap testing [5] |
| WoLF PSORT | Subcellular localization prediction | Protein localization inference [5] |
The architectural blueprint of plant NLR immune receptors—featuring conserved N-terminal signaling domains, central nucleotide-binding switches, and C-terminal ligand-sensing regions—has been maintained throughout land plant evolution while permitting remarkable functional diversification. The classification into CNL, TNL, and RNL subfamilies reflects fundamental divisions in signaling mechanism and evolutionary history. Current evidence suggests that NLR genes undergo continuous birth-death evolution driven by pathogen pressure, resulting in species-specific repertoires optimized for particular ecological niches. The methodological framework presented here enables comprehensive characterization of NLR genes across plant species, facilitating discoveries that bridge evolutionary genomics and molecular immunity. Future research leveraging pan-NLRome studies will further elucidate structure-function relationships in this critical plant immune receptor family, with applications in crop improvement and sustainable agriculture.
The nucleotide-binding domain and leucine-rich repeat (NLR) gene family represents a cornerstone of the innate immune system across the plant kingdom. This in-depth technical review synthesizes current genomic and evolutionary evidence establishing that the core components of NLR immune receptors originated in the common ancestor of green plants approximately one billion years ago. We trace the evolutionary trajectory of NLR genes from early aquatic algae to modern land plants, highlighting key diversification events driven by ecological adaptation. The article provides detailed methodologies for NLR identification and analysis, presents quantitative comparative genomics across species, and details the co-evolution of NLR receptors with downstream signaling components. This synthesis provides a comprehensive framework for researchers and drug development professionals understanding the deep evolutionary history of plant immunity mechanisms.
Plant immunity relies on a sophisticated two-layer system for pathogen detection. The first layer involves pattern-triggered immunity (PTI) mediated by cell surface-localized pattern recognition receptors (PRRs) that detect conserved pathogen-associated molecular patterns (PAMPs). The second layer employs intracellular NLR receptors that recognize pathogen effector proteins, activating effector-triggered immunity (ETI) which often includes a hypersensitive response and programmed cell death [3].
NLR proteins are characterized by a conserved tripartite domain architecture: a central nucleotide-binding site (NBS) domain, a C-terminal leucine-rich repeat (LRR) domain, and variable N-terminal domains that define subclass specificity. In seed plants, three major NLR subclasses have been identified: TNLs (with Toll/Interleukin-1 Receptor domains), CNLs (with Coiled-Coil domains), and RNLs (with Resistance to Powdery Mildew8 domains) [7] [3]. The RNL subclass functions primarily as "helper" NLRs that transduce immune signals from "sensor" CNLs and TNLs [3].
Recent research has revealed that PTI and ETI are not independent systems but work synergistically to enhance plant immune responses [3]. This review examines the evolutionary origins of the NLR system and its coordinated development with signaling pathways throughout plant evolution.
Evolutionary Origins: From Green Algae to Land Plants
Deep Evolutionary Roots
Comprehensive genomic analyses have revealed that the evolutionary assembly of NLR core building blocks occurred in the ancestors of early plants, with traceable origins in green algae (Figure 1) [8] [3]. Although plant and animal NLRs share similar domain architecture, they evolved independently through convergent evolution, with plant NLRs tracing specifically to the common ancestor of all green plants (Viridiplantae) [3].
Table 1: NLR Distribution Across Plant Lineages
| Plant Group | Representative Species | NLR Presence | Subclasses Identified | Key Evolutionary Notes |
|---|---|---|---|---|
| Green Algae | Chara braunii | Limited | Proto-NLRs | Core building blocks assembled |
| Bryophytes | Physcomitrella patens | Present | TIR and non-TIR | Present in early land plants |
| Basal Angiosperms | Amborella trichopoda | Present | TNL and CNL | Both major subclasses present |
| Monocots | Oryza sativa | Present | CNL, RNL | TNLs generally absent |
| Eudicots | Arabidopsis thaliana | Present | TNL, CNL, RNL | All three subclasses present |
The evolutionary transition to land approximately 500 million years ago represented a pivotal moment for NLR expansion. Land plants faced increased pathogen pressure in this new environment, driving remarkable NLR gene diversification. Genomic analyses reveal only a few NLR genes in most charophyte and chlorophyte genomes, contrasting sharply with the dozens to hundreds found in land plant genomes [3]. This expansion coincided with the development of more complex immune signaling networks necessary for terrestrial survival.
Lineage-Specific Diversification and Loss
Following the divergence of major plant lineages, NLR evolution exhibited distinct trajectories characterized by specific patterns of gene expansion, contraction, and loss:
Monocot Evolution: Comprehensive studies across multiple monocot orders (Poales, Zingiberales, Arecales, Asparagales, and Alismatales) have revealed a significant reduction or loss of TNL-type genes [9]. Both experimental and bioinformatic evidence consistently shows absence of TNL sequences in monocots, despite their presence in basal angiosperms and gymnosperms [9].
Eudicot Evolution: In contrast to monocots, eudicots maintained both TNL and CNL subclasses, with numerous examples of functional diversification. Recent evidence shows that some NLRs in eudicots have expanded functions beyond pathogen recognition, including roles in environmental sensing [3].
Ecological Adaptation Drivers: Comparative genomic analyses reveal that NLR gene family size correlates strongly with ecological adaptation rather than phylogenetic position [3]. Species facing diverse pathogen pressures typically maintain expanded and diversified NLR repertoires, demonstrating the dynamic nature of this gene family in response to environmental challenges.
Figure 1. Evolutionary trajectory of NLR genes in plants. The diagram traces the origin of NLR building blocks to green algal ancestors, followed by key diversification events during plant terrestrialization and subsequent lineage-specific evolution.
Methodologies for NLR Gene Identification and Analysis
Genomic Mining and Annotation Pipelines
Accurate identification and annotation of NLR genes present particular challenges due to their complex domain architecture and frequent presence in gene clusters. Several specialized computational approaches have been developed:
Standard NLR Annotation Workflow:
- Domain Identification: Use HMMER3 with Pfam NBS (NB-ARC) domain profile (PF00931) for initial screening (E-value < 10⁻⁴) [7]
- BLAST Validation: Perform BLASTp search against all protein sequences (E-value = 1.0) to identify divergent homologs [7]
- Domain Verification: Confirm NBS domains using hmmscan against local Pfam-A database [7]
- Motif Analysis: Identify conserved motifs using MEME suite and visualize with WebLogo [7]
- Subclassification: Categorize based on N-terminal domains (TIR, CC, RPW8) and conserved kinase-2 motifs [9]
Polyploid Genome Challenges: For complex polyploid genomes like sugarcane, researchers developed DaapNLRSeek, a diploidy-assisted annotation pipeline that combines NLR-Annotator, GeMoMa, and Augustus programs with manual curation [10]. This approach successfully annotated 3,362-7,138 NLR genes across various sugarcane cultivars, dramatically improving upon automated annotations [10].
Phylogenetic and Evolutionary Analysis
Reconstructing NLR evolutionary history requires specialized phylogenetic methods:
- Sequence Alignment: Extract NBS domains and align using ClustalW or MAFFT
- Model Selection: Use ModelFinder to identify best-fit substitution models [7]
- Tree Construction: Perform maximum likelihood analysis with IQ-TREE using SH-aLRT and UFBoot2 branch support (1,000 bootstraps) [7]
- Reconciliation: Compare gene trees with species trees using Notung software to infer duplication/loss events [7]
Table 2: Essential Bioinformatics Tools for NLR Research
| Tool Name | Application | Methodology | Specialized Use Cases |
|---|---|---|---|
| NLR-Annotator | NLR locus identification | Domain-based mining | Standard for diploid genomes |
| DaapNLRSeek | Polyploid NLR annotation | Diploid-assisted pipeline | Complex polyploid genomes [10] |
| NLRtracker | Automated annotation | Homology-based | Well-annotated reference genomes |
| Notung | Gene tree/species tree reconciliation | Duplication/loss inference | Evolutionary analysis [7] |
| MCScanX | Gene duplication typing | Synteny analysis | Genome evolution studies [7] |
Genomic Case Studies: NLR Evolution in Specific Plant Families
Dynamic Evolution in Apiaceae Species
Comparative genomic analysis of four Apiaceae species (Angelica sinensis, Coriandrum sativum, Apium graveolens, and Daucus carota) reveals dynamic NLR evolution in this medically and agriculturally important family [7]. The number of NLR genes varies significantly, ranging from 95 in A. sinensis to 183 in C. sativum (Table 3).
Table 3: NLR Gene Composition in Apiaceae Species
| Species | Total NLR Genes | CNL Subclass | TNL Subclass | RNL Subclass | Evolutionary Pattern |
|---|---|---|---|---|---|
| Angelica sinensis | 95 | 74 (77.9%) | 12 (12.6%) | 9 (9.5%) | Contraction after expansion |
| Coriandrum sativum | 183 | 147 (80.3%) | 22 (12.0%) | 14 (7.7%) | Contraction after expansion |
| Apium graveolens | 153 | 118 (77.1%) | 21 (13.7%) | 14 (9.2%) | Contraction after expansion |
| Daucus carota | 149 | 119 (79.9%) | 16 (10.7%) | 14 (9.4%) | Contraction pattern |
Phylogenetic analysis of these NLR genes indicates they were derived from approximately 183 ancestral NLR lineages in the Apiaceae common ancestor, with differential gene loss and gain events during speciation [7]. The study demonstrated that D. carota experienced a contraction pattern of ancestral NLR lineages, while the other three species showed a pattern of contraction following initial expansion [7].
NLR Conservation and Divergence in Cereal Crops
The Poaceae family (grasses) provides excellent examples of lineage-specific NLR evolution. The near-complete absence of TNL genes in monocots represents a major evolutionary divergence from eudicots [9]. Despite this subclass loss, cereal crops have maintained robust immune systems through:
- Expanded CNL Repertoires: Diversification of CNL genes to compensate for TNL absence
- RNL Helper Specialization: Co-evolution of RNL genes with specific signaling pathways
- Paired NLR Networks: Emergence of genetically linked NLR pairs functioning together
Recent research has identified functionally paired NLR genes in wheat, such as Pm68-1 and Pm68-2, which confer resistance to powdery mildew [11]. Transgenic assays demonstrate that neither gene alone provides resistance, but together they activate a robust immune response, highlighting the sophisticated coordination that has evolved in cereal NLR systems [11].
Signaling Pathways and Co-evolution with Immune Components
NLR Activation Mechanisms
Plant NLRs have evolved distinct activation mechanisms that define their immune functions:
Sensor NLRs (CNLs and TNLs):
- Direct Activation: Some CNLs (e.g., ZAR1 in Arabidopsis) oligomerize to form calcium-permeable channels themselves, initiating downstream signaling [3]
- Helper-Dependent: Other sensor NLRs require helper NLRs (RNLs) to transduce immune signals [3]
Helper NLRs (RNLs):
- Function as signaling hubs that amplify immune responses from sensor NLRs
- Typically form plasma membrane channels that facilitate calcium influx [3]
- Essential for TNL signaling through the EDS1-PAD4-ADR1 module [3]
Co-evolution with Downstream Components
NLR receptors have co-evolved with key signaling components, creating integrated immune networks:
- EDS1 Family Evolution: The origin and divergence of the EDS1 gene family significantly reshaped the ETI system in seed plants, particularly for TNL signaling [3]
- NRC Network: Some CNLs require NRC (NLR-required for cell death) proteins for signal transduction [3]
- Calcium Signaling: Coordination with calcium-dependent protein kinases creates amplification loops
Figure 2. NLR immune signaling network. The diagram illustrates the coordinated relationships between sensor NLRs, helper NLRs, and downstream signaling components in activating plant immunity.
Research Reagent Solutions and Experimental Applications
The study of NLR evolution and function requires specialized research tools and reagents. The following table summarizes essential resources for investigators in this field:
Table 4: Essential Research Reagents for NLR Studies
| Reagent/Resource | Function/Application | Technical Specifications | Research Context |
|---|---|---|---|
| NLR-Annotator | Genome-wide NLR identification | Domain-based HMM profiling | Standard for diploid genomes [10] |
| DaapNLRSeek Pipeline | Polyploid NLR annotation | Diploid-assisted annotation | Complex sugarcane genomes [10] |
| Nicotiana benthamiana | Transient expression system | HR cell death assays | Functional validation [10] [11] |
| PacBio HiFi Sequencing | NLR allele resolution | Long-read technology | Haplotype characterization [11] |
| GeMoMa | Homology-based annotation | Gene model prediction | Integration in DaapNLRSeek [10] |
| Augustus | Ab initio gene prediction | Species-specific parameters | NLR gene modeling [10] |
These research tools have enabled significant advances in NLR biology, including the identification of two sugarcane paired NLRs that induce immune responses in Nicotiana benthamiana [10] and the characterization of the Pm68 NLR pair in wheat that confers powdery mildew resistance without significant agronomic trade-offs [11].
The evolutionary history of NLR genes represents a remarkable case study in adaptive evolution and immune system specialization. From their origin in the common ancestor of green plants to their lineage-specific diversification, NLR genes have continually evolved to meet changing pathogenic challenges. The core building blocks assembled in aquatic algae provided the foundation for the sophisticated immune systems that enabled plant terrestrialization and subsequent diversification.
Future research directions should focus on:
- Understanding NLR functional integration with other immune receptors
- Exploring NLR evolution in underrepresented plant lineages
- Harnessing evolutionary insights for crop improvement through NLR engineering
- Investigating how NLR diversification correlates with habitat-specific pathogen pressures
The deep evolutionary history of NLR genes, traced through modern genomic analyses, provides not only insights into plant adaptation but also practical knowledge for developing durable disease resistance in agricultural systems. As genomic technologies continue to advance, our understanding of NLR evolution will undoubtedly reveal additional layers of complexity in these essential components of the plant immune system.
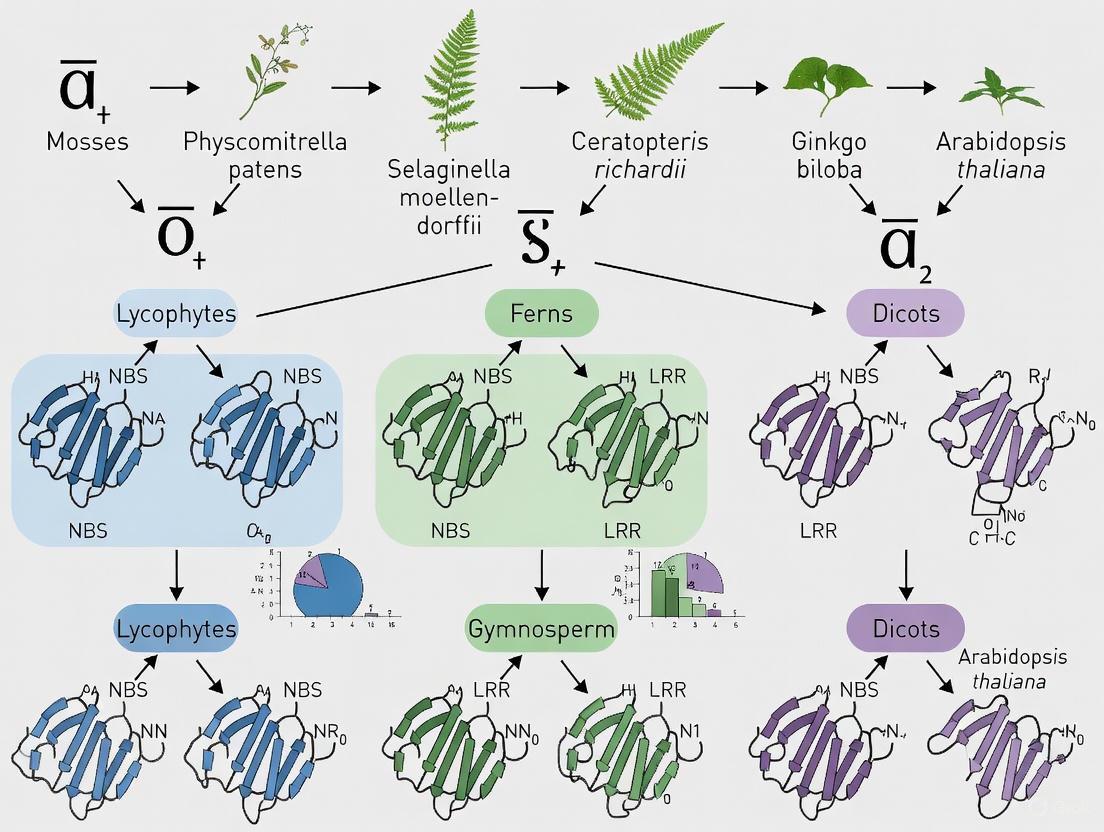
The nucleotide-binding domain and leucine-rich repeat receptors (NLRs) constitute a major component of the plant innate immune system, exhibiting extraordinary diversification across flowering plants. This review examines the minimal NLR repertoire of the moss Physcomitrella patens as an evolutionary snapshot of the ancestral land plant immune system. With approximately 25 NLR genes, P. patens provides a crucial phylogenetic reference point for understanding the expansion, diversification, and functional specialization of NLRs in vascular plants. We synthesize current knowledge on the architectural features, taxonomic distribution, and experimental methodologies relevant to studying NLRs in this model bryophyte, contextualizing these findings within the broader evolutionary trajectory of plant immunity from early land plants to modern angiosperms.
Plants rely entirely on innate immunity systems to defend against pathogens, utilizing intracellular NLR proteins as key receptors for detecting pathogen-derived effectors and initiating effector-triggered immunity (ETI) [1]. NLRs are modular proteins characterized by three core building blocks: a central nucleotide-binding (NB-ARC) domain, C-terminal leucine-rich repeats (LRRs), and variable N-terminal domains that define major NLR subclasses [1] [3].
The NLR family has undergone massive expansion in flowering plants, with genomes encoding dozens to hundreds of NLR genes [1]. This expansion contrasts sharply with the minimal NLR complement in bryophytes, positioning P. patens as a critical model for understanding the evolutionary foundations of plant immunity. As a descendant of one of the earliest land plant lineages, P. patens offers unique insights into the primordial NLR repertoire before its extensive diversification in vascular plants [1] [12].
The NLR Repertoire ofPhyscomitrella patensin Comparative Context
Quantitative Analysis of NLR Distribution Across Plant Lineages
Table 1: NLR Gene Repertoire Size Across Representative Plant Species
| Species | Common Name | Classification | Total NLRs | TNLs | CNLs | XNLs | Reference |
|---|---|---|---|---|---|---|---|
| Physcomitrella patens | Moss | Bryophyte | 25 | 8 | 9 | 8 | [1] |
| Selaginella moellendorffii | Spike moss | Lycophyte | 2 | 0 | NA | NA | [1] |
| Arabidopsis thaliana | Thale cress | Dicot | 151 | 94 | 55 | 0 | [1] |
| Oryza sativa | Rice | Monocot | 458 | 0 | 274 | 182 | [1] |
| Vitis vinifera | Wine grape | Dicot | 459 | 97 | 215 | 147 | [1] |
| Zea mays | Maize | Monocot | 95 | 0 | 71 | 23 | [1] |
The NLR repertoire in P. patens is remarkably compact, with only 25 NLR genes identified in its genome [1]. This minimal complement includes 8 TIR-type NLRs (TNLs), 9 CC-type NLRs (CNLs), and 8 NLRs with atypical N-terminal domains (XNLs) [1]. This stands in stark contrast to the hundreds of NLRs found in many angiosperm genomes, highlighting the extensive expansion that occurred during vascular plant evolution.
The small NLR repertoire in P. patens is particularly significant when considering the species' genomic characteristics. P. patens has a genome size of approximately 511 Mbp and exhibits a unique architecture lacking the TE-rich pericentromeric and gene-rich distal regions typical of flowering plant genomes [12]. Despite undergoing two rounds of whole genome duplication (WGD) – events often associated with gene family expansion – the NLR family remains notably constrained in this moss [12].
Phylogenetic Distribution and Evolutionary History
Comparative analyses reveal that NLRs originated in green algae and were already well-established in the common ancestor of land plants [13]. The minimal NLR repertoire in P. patens, along with the even more reduced complement in the lycophyte Selaginella moellendorffii (2 NLRs), suggests dynamic patterns of NLR expansion and contraction throughout plant evolution [1].
Notably, P. patens possesses both major NLR subfamilies (TNLs and CNLs), indicating that the divergence between these lineages predates the separation of bryophytes from vascular plants [1] [3]. This contrasts with monocots, which typically lack TNLs entirely, suggesting secondary loss in this lineage [1]. The presence of diverse XNLs in P. patens further indicates early experimentation with different N-terminal domain architectures in NLR proteins [1].
Methodological Framework for NLR Research inPhyscomitrella patens
Genomic and Transcriptomic Workflows
Figure 1: Experimental Workflow for NLR Gene Identification and Characterization in P. patens
The experimental workflow for NLR characterization in P. patens begins with comprehensive genome sequencing and transcriptome assembly to establish a reference for NLR identification [12]. The P. patens genome assembly provides a chromosome-scale resource that enables precise genomic context analysis of NLR genes, including their association with transposable elements and duplication history [12].
NLR identification typically employs domain-based search pipelines using conserved protein domains, particularly the NB-ARC domain (Pfam PF00931), as bait for retrieving NLR sequences from genomic and transcriptomic datasets [14] [15]. Following identification, domain architecture analysis classifies NLRs into subfamilies based on N-terminal domains (TIR, CC, or other), while phylogenetic analysis reconstructs evolutionary relationships among NLR sequences within and across species [1] [13].
Essential Research Tools and Reagents
Table 2: Essential Research Reagents and Resources for NLR Studies in P. patens
| Category | Specific Resource | Application in NLR Research | Key Features |
|---|---|---|---|
| Genomic Resources | P. patens chromosome-scale assembly | Genomic context analysis of NLR genes | 511 Mbp genome, 27 chromosomes [12] |
| Identification Tools | NLRtracker pipeline | Genome-wide NLR identification | Domain-based annotation of NLR genes [16] |
| Domain Databases | Pfam NB-ARC domain (PF00931) | NLR sequence identification | Curated hidden Markov models for NLR detection [14] |
| Expression Data | RNA-seq datasets | Expression profiling of NLR genes | Tissue-specific, stress-responsive expression patterns [16] |
| Genetic Tools | Targeted gene knockout system | Functional validation of NLR genes | Efficient homologous recombination in P. patens [17] |
| Comparative Data | Plant NLR repertoires | Evolutionary analysis | Cross-species comparisons of NLR architecture [1] [14] |
The unique genetic transformation system of P. patens, which allows highly efficient targeted gene knockout via homologous recombination, provides a powerful functional validation tool not readily available in most plant models [17]. This capability enables direct testing of NLR gene functions through reverse genetics approaches.
Additionally, comparative genomic frameworks have been developed specifically for analyzing NLR evolution across multiple species. These include pipelines for identifying NLRs with integrated domains (NLR-IDs), which represent fusions between NLRs and other protein domains that can serve as pathogen effector baits [14] [15]. While such integrated domains appear more prevalent in angiosperms, their detection in bryophyte genomes provides insights into the early evolution of these composite immune receptors.
Evolutionary Implications of the Minimal Bryophyte NLR Repertoire
Insights into NLR Origin and Diversification
The compact NLR repertoire in P. patens provides critical insights into the early evolution of plant immune systems. Several lines of evidence suggest that plant and animal NLRs arose independently despite their similar architecture and function [1]. The distinct nature of the plant NB-ARC domain compared to the animal NACHT domain supports this independent origin [1].
The presence of a diverse but small NLR repertoire in P. patens indicates that the fundamental NLR architecture was already established in the earliest land plants but had not undergone the dramatic expansion characteristic of flowering plants [1] [3]. This expansion in angiosperms is thought to be driven by ecological adaptation to diverse pathogen pressures [3], with lineage-specific expansions and contractions reflecting particular pathogenic challenges.
Figure 2: Evolutionary Relationships of NLR Subfamilies Across Land Plants
The evolutionary trajectory of NLR genes reveals several significant patterns. First, the TNL and CNL subfamilies diverged early in land plant evolution, with both present in P. patens [1] [3]. Second, the RNL subfamily (RPW8-NLRs) emerged later, likely in vascular plants, as these are absent from P. patens but present in conifers and angiosperms [3] [13]. Third, different plant lineages have experienced differential expansion of NLR subfamilies, with monocots losing TNLs entirely while expanding CNLs and RNLs [1] [13].
Functional and Structural Conservation
Despite the numerical expansion in angiosperms, studies indicate remarkable functional conservation of NLR signaling mechanisms across land plants. The demonstration of interfamily transfer of NLR functions from their original species to phylogenetically distant species implies evolutionary conservation of the underlying immune mechanisms [1]. This functional conservation suggests that the core signaling mechanisms established in early land plants, like P. patens, have been largely maintained throughout plant evolution.
Structural analyses of NLR proteins reveal conserved motifs within the NB-ARC domain, including the P-loop, kinase motifs, RNBS, GLPL, and MHD motifs, which are involved in nucleotide binding and conformational changes during activation [13]. These motifs show conservation between P. patens and angiosperm NLRs, indicating maintenance of core biochemical functions across land plant evolution.
The minimal NLR repertoire of Physcomitrella patens provides an evolutionary snapshot of the foundational elements of the plant immune system before its extensive diversification in vascular plants. With approximately 25 NLR genes representing major NLR subfamilies, P. patens offers a simplified system for understanding the core principles of NLR-mediated immunity without the complexity of massively expanded gene families found in angiosperms.
Future research directions should include functional characterization of individual P. patens NLRs using its efficient gene targeting system, comparative analyses of NLR architectures across bryophyte species, and structural studies of bryophyte NLRs to understand conserved activation mechanisms. Such approaches will continue to illuminate how this ancient immune receptor family has evolved to meet diverse pathogenic challenges across the plant kingdom.
The study of P. patens NLRs not only reveals the evolutionary history of plant immunity but may also provide insights for engineering disease resistance in crops by identifying conserved functional principles that can be transferred across plant lineages. As genomic resources for non-seed plants continue to expand, our understanding of NLR evolution will become increasingly refined, further highlighting the value of bryophytes as evolutionary snapshots of primordial plant immune systems.
In plants, which lack an adaptive immune system, Nucleotide-binding domain and Leucine-rich Repeat (NLR) proteins serve as critical intracellular immune receptors that detect pathogen effectors and initiate robust defense responses, a mechanism known as effector-triggered immunity (ETI) [1]. NLR genes constitute one of the largest and most variable gene families in the plant kingdom, with extraordinary sequence, structural, and regulatory diversity enabling recognition of rapidly evolving pathogens [18]. Despite similar architecture and function to animal NLRs, comparative genomic analyses reveal that plant NLRs arose independently during evolution, suggesting convergent evolutionary solutions to pathogen detection [1]. The dramatic expansion of NLR gene families in flowering plants, particularly in agricultural crops, represents an evolutionary arms race with pathogens that has shaped plant genomes and provided crucial resources for breeding disease-resistant crops. This review examines the patterns, mechanisms, and functional consequences of NLR proliferation across land plants, with specific emphasis on the contrasting evolutionary trajectories observed from ancient mosses to modern crops like wheat and grapevine.
Evolutionary History and Comparative Genomics of NLR Repertoires
Lineage-Specific Expansion Patterns
The NLR family has undergone massive expansion in numerous flowering plant species, creating one of the largest and most variable plant protein families [1]. Genomic surveys reveal striking differences in NLR copy numbers across plant lineages, with particularly dramatic expansions observed in flowering plants compared to non-vascular plants [1] [19].
Table 1: NLR Gene Repertoire Across Representative Plant Species
| Species | Common Name | Genome Size (Mbp) | Total NLRs | TNLs | CNLs | XNLs | Reference |
|---|---|---|---|---|---|---|---|
| Selaginella moellendorffii | Spike moss | 100 | 2 | 0 | NA | NA | Yue et al. [1] |
| Physcomitrella patens | Moss | 511 | 25 | 8 | 9 | 8 | Xue et al. [1] |
| Arabidopsis thaliana | Thale cress | 125 | 151 | 94 | 55 | 0 | Meyers et al. [1] |
| Carica papaya | Papaya | 372 | 34 | 6 | 4 | 1 | Porter et al. [1] |
| Vitis vinifera | Wine grape | 487 | 459 | 97 | 215 | 147 | Yang et al. [1] |
| Oryza sativa | Rice | 466 | 458 | 0 | 274 | 182 | Li et al. [1] |
| Zea mays | Maize | 2400 | 95 | 0 | 71 | 23 | Li et al. [1] |
| Triticum aestivum | Bread wheat | ~17,000 | ~3,400 | NA | NA | NA | Walkowiak et al. [20] |
| Haynaldia villosa | Wild grass | NA | 1,320 | NA | NA | NA | Cheng et al. [20] |
The data reveals several key evolutionary patterns. First, ancestral land plants like the spike moss Selaginella moellendorffii and moss Physcomitrella patens possess minimal NLR repertoires (approximately 2 and 25 NLRs, respectively), suggesting limited NLR diversification before the emergence of vascular plants [1]. Second, flowering plants exhibit tremendous variation in NLR copy numbers without clear phylogenetic correlation, indicating species-specific expansion and contraction events [1]. For instance, while papaya contains only 34 NLRs, grapevine boasts 459 NLRs despite similar genome sizes [1]. Third, certain monocots like rice have undergone substantial NLR expansion (458 NLRs), while others like maize maintain relatively modest repertoires (95 NLRs) despite massive genome sizes [1]. Finally, polyploid species like bread wheat contain exceptionally large NLR complements (~3,400 NLRs), suggesting that genome duplication events provide raw genetic material for NLR diversification [20].
Evolutionary Drivers of NLR Contraction and Expansion
Recent pangenomic studies reveal that NLR evolution involves rapid gene loss and gain, with copy numbers differing up to 66-fold among closely related species [21]. Several evolutionary patterns have emerged linking NLR repertoire dynamics to ecological factors:
- Ecological Specialization: NLR contraction is strongly associated with adaptations to aquatic, parasitic, and carnivorous lifestyles [21]. The convergent NLR reduction in aquatic plants resembles the limited NLR expansion observed in green algae before terrestrial colonization, suggesting that certain ecological niches reduce selective pressure from soil-borne pathogens [21].
- Co-evolution with Signaling Components: A co-evolutionary pattern exists between NLR subclasses and plant immune pathway components. For example, TNL loss appears associated with deficiencies in the EDS1–SAG101–NRG1 signaling module, suggesting integrated evolution of receptors and downstream signaling components [21].
- Dynamic Lineage-Specific Patterns: Comparative genomic analysis of Apiaceae species reveals varying evolutionary patterns, with Daucus carota showing contraction of ancestral NLR lineages, while Angelica sinensis, Coriandrum sativum, and Apium graveolens exhibit initial expansion followed by contraction [7]. These findings suggest that rapid and dynamic gene content variation has shaped NLR evolutionary history across plant families.
Diagram 1: Evolutionary dynamics shaping NLR repertoires. NLR repertoires undergo expansion through whole genome duplication (WGD) and small-scale duplications (SSD), while contraction occurs via gene loss, ecological specialization, and co-evolution with signaling components.
Genomic Architecture and Mechanisms of NLR Diversification
Genomic Organization and Complex Loci
NLR genes display non-random genomic distribution, frequently organizing into complex multi-loci regions with allelic series ranging from moderate to extreme sequence divergence [18]. Pangenomic studies in Arabidopsis thaliana have defined 121 pangenomic NLR neighborhoods that vary substantially in size, content, and complexity [18]. These neighborhoods represent genomic regions with concentrated NLR density, suggesting preferential retention or expansion in specific chromosomal locations.
Two primary genomic architectures characterize NLR organization:
- Singleton loci: Individual NLR genes spatially separated from other NLRs by at least 250 kb [7].
- Clustered loci: Multiple NLR genes located within 250 kb of each other, often exhibiting sequence homology and functional relatedness [7].
Research demonstrates that distinct evolutionary processes act on NLR neighborhoods defending against different pathogen types, with biotrophic pathogens exerting unique selective pressures that shape NLR architecture [18]. The increased complexity in NLR neighborhoods centers specifically on NLRs themselves rather than surrounding genomic regions, highlighting the targeted nature of diversification mechanisms [18].
Molecular Mechanisms Generating NLR Diversity
Plant genomes employ multiple uncorrelated mutational and genomic processes to generate NLR diversity, including:
- Tandem Duplications: Local gene duplications create arrays of homologous NLR genes that subsequently diverge in sequence and function [19]. These tandem arrays facilitate the emergence of novel pathogen recognition specificities through ectopic recombination and gene conversion events.
- Whole Genome Duplication (WGD): Polyploidization events provide duplicated NLR loci that escape functional constraints and accumulate substitutions [19]. The correlation between ploidy level and NLR number in species like wheat and apple supports the importance of WGD in NLR expansion [20].
- Transposable Element Activity: TEs contribute to NLR diversification by causing insertional polymorphisms in regulatory regions, affecting gene expression, and facilitating domain shuffling [22]. TE insertions near NLR genes can create epigenetic variation that influences expression patterns.
- Integrated Domains (IDs): Many NLRs incorporate novel protein domains through exon duplication or ectopic recombination, creating NLR-IDs that can function as integrated decoys for pathogen effectors [23]. Kinases, WRKY domains, and zinc-finger BED domains represent some of the most frequently integrated domains [23] [20].
Table 2: Major Types of Integrated Domains in NLR Proteins
| Integrated Domain | Frequency | Proposed Function | Example NLRs |
|---|---|---|---|
| Kinase domains | High | Phosphorylation signaling | Multiple rice NLRs |
| WRKY domains | High | Transcription regulation | RRS1/RPS4 (Arabidopsis) |
| BED zinc fingers | High | DNA binding | Yr5, Yr7 (wheat); Xa1 (rice) |
| Heavy Metal-Associated (HMA) | Medium | Effector binding | RGA5 (rice) |
| Kelch domains | Variable | Protein-protein interaction | Expanded in Haynaldia villosa [20] |
| DUF948 | Low | Unknown function | Unique to Haynaldia villosa [20] |
Research Methodologies for NLR Gene Identification and Characterization
Advanced Genomic Approaches for NLR Discovery
Comprehensive identification of NLR genes presents significant challenges due to their large numbers, sequence diversity, and complex genomic organization. Several specialized methodologies have been developed to address these challenges:
SMRT-RenSeq (Single-Molecule Real-Time Resistance Gene Enrichment Sequencing): This powerful method combines targeted capture of NLR genes using biotinylated baits representing conserved NLR domains with long-read sequencing technologies [20]. The approach enables selective sequencing of full-length NLRs, even from species lacking reference genomes. Application in the wild wheat relative Haynaldia villosa identified 1,320 NLRs, including 772 complete NLR genes, revealing exceptional NLR diversity in wild species [20].
NLR-Annotator Pipeline: Specialized bioinformatic tools have been developed specifically for NLR identification from genomic sequences. NLR-Annotator improves upon previous tools by accurately distinguishing borders between different NLRs located in long contigs, enabling more precise annotation of clustered NLR loci [20].
Pangenome Graph Approaches: Advanced pangenomic frameworks enable nuanced analysis of NLR evolution in genomic context by integrating genome-specific full-length transcript, homology, and transposable element information [18]. This approach revealed that NLR diversity arises from multiple uncorrelated mutational and genomic processes requiring multiple metrics to fully capture NLR variation [18].
Functional Validation Techniques
Determining the function of identified NLR genes requires specialized experimental approaches:
Virus-Induced Gene Silencing (VIGS): This technique uses modified viruses to deliver sequences that trigger degradation of target NLR transcripts, enabling functional analysis through knock-down phenotypes. Silencing of GaNBS (OG2) in resistant cotton demonstrated its putative role in virus resistance [19].
Heterologous Expression Systems: Transient expression in model systems like Nicotiana benthamiana allows testing of NLR function across species boundaries. For example, nuclear localization signals in Yr7 were functionally validated through truncated versions expressed in N. benthamiana [23].
Protein Interaction Studies: Protein-ligand and protein-protein interaction assays reveal interaction networks between NLRs and pathogen components. Studies demonstrated strong interaction of putative NBS proteins with ADP/ATP and different core proteins of the cotton leaf curl disease virus [19].
Diagram 2: NLR identification and validation workflow. The process begins with NLR enrichment and long-read sequencing, followed by specialized annotation, and concludes with multiple validation approaches to confirm function.
Table 3: Essential Research Reagents for NLR Studies
| Reagent/Resource | Function/Application | Key Features | Example Use |
|---|---|---|---|
| SMRT-RenSeq Baits | Enrichment of NLR sequences | ~80% sequence identity sufficient for capture | NLR identification in species without reference genomes [20] |
| NLR-Annotator | Bioinformatics annotation | Distinguishes borders between adjacent NLRs | Genome-wide NLR annotation [20] |
| ANNA Database (Angiosperm NLR Atlas) | Comparative genomics | >90,000 NLR genes from 304 angiosperm genomes | Evolutionary studies of NLR contraction/expansion [21] |
| Tissue Culture Materials | Plant transformation | Species-specific protocols | Functional validation in crop plants |
| VIGS Vectors | Virus-Induced Gene Silencing | Gene function analysis through knock-down | Validating NLR function in cotton [19] |
| Nicotiana benthamiana | Heterologous expression system | Permissive for NLR signaling across species | Testing NLR function and localization [23] |
Case Studies: NLR Expansion in Wheat and Grapevine
Exceptional NLR Proliferation in Wheat and Wild Relatives
The tribe Triticeae, including wheat and its wild relatives, exhibits extraordinary NLR expansion, representing some of the largest NLR repertoires documented in plants:
Bread Wheat (Triticum aestivum): Hexaploid bread wheat contains approximately 3,400 NLR genes, the largest number reported thus far in any plant species [20]. This massive expansion results from the combination of polyploidization (merging three genomes) and subsequent diversification events.
Wild Relatives: Haynaldia villosa, a wild diploid wheat relative with proven potential for wheat improvement, possesses 1,320 NLR genes despite its diploid status [20]. SMRT-RenSeq analysis revealed 15 types of integrated domains in 52 NLRs, with Kelch and B3 NLR-IDs showing particular expansion, while DUF948, NAM-associated and PRT_C domains were detected as unique integrations [20].
Genomic Architecture: NLRs in wheat and related grasses show perfect homoeologous relationships with group 1, 2, 3, 5, and 6 chromosomes in other Triticeae species. However, NLRs physically located on chromosome 4VL in H. villosa were largely predicted to reside on homoeologous group 7, suggesting chromosomal repatterning [20].
NLR Expansion in Grapevine Genomes
Comparative analysis of cultivated and wild grapevine genomes reveals distinctive patterns of NLR evolution:
Cultivated vs. Wild Grapevines: Wild North American grapevine species, including Vitis labrusca, exhibit large expansions of NLR genes compared to cultivated European grapevines [22]. This expansion may contribute to their superior disease resistance profiles.
Associations with Breeding History: The extensive use of wild grapevine species as rootstocks to combat phylloxera infestation in European vineyards demonstrates the practical application of NLR diversity from wild species for sustainable agriculture [22].
Heterozygosity and Structural Variation: Cultivated grapevine genomes display approximately twice the heterozygosity of wild grapevine genomes [22]. Approximately 30% of V. labrusca and 48% of V. vinifera Chardonnay genes were heterozygous or hemizygous, with considerable variation in gene zygosity between collinear genes in Chardonnay and V. labrusca [22].
The "great expansion" of NLR genes in flowering plants represents a remarkable evolutionary adaptation to pathogen pressure. From minimal repertoires in ancestral lineages like mosses and spike mosses to massive collections of several thousand NLRs in crops like wheat, this proliferation demonstrates the dynamic nature of plant genomes in response to biotic challenges. The differential retention and expansion of NLRs between closely related species, and between wild and cultivated forms, highlights the complex interplay between ecological factors, genome dynamics, and pathogen pressures.
Future research directions should focus on several key areas: (1) understanding how NLR diversity translates to functional diversity in pathogen recognition; (2) elucidating the signaling networks that connect expanded NLR repertoires to defense outputs; (3) harnessing wild NLR diversity for crop improvement through advanced breeding technologies; and (4) exploring the potential fitness costs associated with maintaining large NLR repertoires and how plants mitigate these costs. As genomic technologies continue to advance, particularly in pangenome construction and single-cell omics, our understanding of NLR expansion and evolution will undoubtedly deepen, providing new insights into plant immunity and new tools for sustainable agriculture.
The evolutionary history of land plants is marked by a constant arms race with a diverse array of pathogens. Central to this billion-year conflict are nucleotide-binding site leucine-rich repeat (NLR) proteins, which serve as major intracellular immune receptors responsible for effector-triggered immunity (ETI) in plants [3]. Unlike vertebrates that employ an adaptive immune system with specialized cells, plants rely entirely on this innate immune system, making the diversity and evolution of NLR genes critical for survival [1]. The remarkable structural and functional diversification of NLR genes across land plants, from mosses to dicots, represents a complex evolutionary response to pathogen pressure and ecological adaptation. This review examines the drivers of NLR diversity within the context of plant evolution, synthesizing recent genomic evidence that reveals how pathogen interactions and ecological niches have shaped the expansion, contraction, and functional specialization of this crucial gene family.
NLR Gene Evolution Across Land Plants
Evolutionary Origins and Phylogenetic Distribution
NLR genes originated approximately one billion years ago, coinciding with the emergence of green plants on Earth [3]. The core NLR architecture consists of three fundamental domains: a central nucleotide-binding site (NBS) domain, a C-terminal leucine-rich repeats (LRR) domain, and variable N-terminal domains that define major NLR subclasses [1]. Plant NLRs are categorized into TIR-NLRs (TNLs) with Toll/interleukin-1 receptor domains, CC-NLRs (CNLs) with coiled-coil domains, and RPW8-NLRs (RNLs) with Resistance to powdery mildew8 domains [3].
Phylogenetic analyses reveal striking differences in NLR repertoires across plant lineages. Early land plants such as the bryophyte Physcomitrella patens and the lycophyte Selaginella moellendorffii possess relatively small NLR repertoires of approximately 25 and 2 genes respectively [1]. This modest number stands in stark contrast to the massively expanded NLR families observed in flowering plants, where repertoire sizes can exceed 450 genes, as documented in wine grape (Vitis vinifera) [1]. This pattern indicates that substantial gene expansion occurred primarily after the divergence of flowering plants, likely driven by increased selective pressure from co-evolving pathogens in diverse ecological niches.
Table 1: NLR Repertoire Size Variation Across Plant Lineages
| Species | Common Name | Lineage | Total NLRs | TNLs | CNLs | Other NLRs |
|---|---|---|---|---|---|---|
| Physcomitrella patens | Moss | Bryophyte | 25 | 8 | 9 | 8 |
| Selaginella moellendorffii | Spike Moss | Lycophyte | 2 | 0 | N/A | N/A |
| Arabidopsis thaliana | Thale Cress | Eudicot | 151 | 94 | 55 | 0 |
| Oryza sativa | Rice | Monocot | 458 | 0 | 274 | 182 |
| Vitis vinifera | Wine Grape | Eudicot | 459 | 97 | 215 | 147 |
| Zea mays | Maize | Monocot | 95 | 0 | 71 | 23 |
Genomic Drivers of NLR Diversity
The expansion of NLR genes in flowering plants has been facilitated by several genomic mechanisms, with whole-genome duplication (WGD) and small-scale duplications (SSD) serving as primary drivers [19]. Small-scale duplications include tandem, segmental, and transposon-mediated duplications, which contribute significantly to the generation of NLR diversity [19]. Interestingly, gene families evolving through WGDs seldom undergo SSD events, suggesting distinct evolutionary paths for NLR expansion [19].
A comparative analysis of 34 plant species identified 12,820 NBS-domain-containing genes classified into 168 distinct architectural classes, revealing both classical domain patterns (NBS, NBS-LRR, TIR-NBS, TIR-NBS-LRR) and species-specific structural patterns (TIR-NBS-TIR-Cupin1-Cupin1, TIR-NBS-Prenyltransf, Sugar_tr-NBS) [19]. This extraordinary diversity in domain architecture enables plants to recognize a vast array of pathogen effectors through direct or indirect recognition mechanisms [1].
Recent pangenome studies in Arabidopsis thaliana have further illuminated the extent of intraspecific NLR evolution, with 3,789 NLRs identified across 17 diverse accessions [24]. These NLRs are organized into 121 pangenomic neighborhoods that vary considerably in size, content, and complexity, highlighting the dynamic nature of NLR evolution even within species [24].
Pathogen Pressure as a Driver of NLR Evolution
Co-evolutionary Dynamics with Pathogens
The plant immune system operates on a principle of continuous co-evolution with pathogens, often described by the Red Queen Hypothesis, where plants and pathogens engage in constant adaptation and counter-adaptation [25]. This evolutionary arms race creates frequency-dependent selection that maintains genetic diversity at NLR loci, as rare resistance alleles gain selective advantage against common pathogen strains [25].
Pathogen pressure drives NLR evolution through several molecular mechanisms. Plant NLRs utilize either direct recognition (physical interaction with pathogen effectors) or indirect recognition (sensing modifications of host proteins caused by effectors) [1]. The indirect recognition mechanism enables a single NLR to recognize multiple effectors irrespective of their structures, provided these effectors target the same host protein [1]. Recent evidence demonstrates that some NLRs can also detect multiple sequence-unrelated effectors through direct binding, expanding their recognition capacity [1].
Serial passage experiments with the fungal pathogen Stemphylium solani on clover hosts have demonstrated that pathogens can rapidly adapt to novel hosts within just four generations, showing increased infection rates [25]. This rapid pathogen evolution necessitates corresponding diversification in the host NLR repertoire, creating continuous selective pressure for innovation in immune recognition.
Diversity Generation Mechanisms
Plants have evolved multiple strategies to generate the diversity necessary for recognizing rapidly evolving pathogens. Tandem duplications of NLR genes create genomic arrays that serve as factories for new recognition specificities through ectopic recombination and gene conversion [19]. These mechanisms allow for the shuffling of protein domains, particularly in the LRR region responsible for effector recognition, generating novel binding specificities.
Transposable elements also contribute significantly to NLR diversity, both through their role in gene duplication and by serving as regulatory elements that influence NLR expression [24]. The integration of transposable elements near NLR genes can create novel regulatory contexts and expression patterns, adding another layer of diversity to plant immune responses.
MicroRNAs represent an additional mechanism for regulating NLR diversity. Recent research has revealed that numerous microRNAs target nucleotide sequences encoding conserved motifs within NLRs, including the P-loop, across many flowering plants [19] [1]. This bulk control of NLR transcripts may enable plant species to maintain extensive NLR repertoires without exhausting functional NLR loci, as microRNA-mediated transcriptional suppression could compensate for fitness costs associated with NLR maintenance [19] [1].
Diagram 1: Pathogen-driven NLR evolution. Pathogen pressure activates genomic mechanisms that generate NLR diversity through multiple pathways, creating diverse repertoires that in turn influence pathogen evolution.
Ecological Adaptation and Life History Strategies
Annual vs. Perennial Life History Strategies
The transition between annual and perennial life history strategies has profoundly influenced NLR gene evolution, as demonstrated by comparative genomic studies in the genus Glycine [26]. Annual species, including cultivated soybean (Glycine max) and its wild ancestor (Glycine soja), exhibit expanded NLRomes compared to perennial relatives [26]. Evolutionary timescale analysis indicates that this expansion resulted from recent accelerated gene duplication events between 0.1 and 0.5 million years ago, driven predominantly by lineage-specific and terminal duplications [26].
In contrast, perennial Glycine species experienced significant NLRome contraction following the Glycine-specific whole-genome duplication event approximately 10 million years ago [26]. Despite this overall reduction, perennial lineages have maintained a unique and highly diversified NLR repertoire with limited interspecies synteny [26]. Investigation of gene gain and loss ratios revealed that this diversification resulted from the birth of novel genes following individual speciation events, with G. latifolia exhibiting the highest ratio of novel genes in the tertiary gene pool [26].
Table 2: NLR Evolution in Annual vs. Perennial Glycine Species
| Evolutionary Feature | Annual Species | Perennial Species |
|---|---|---|
| NLRome Size | Expanded | Contracted |
| Major Evolutionary Mechanism | Recent gene duplications (0.1-0.5 MYA) | Birth of novel genes post-speciation |
| Genomic Architecture | Lineage-specific and terminal duplications | Limited interspecies synteny |
| Diversification Pattern | Quantitative expansion | Qualitative diversification |
| Notable Species | G. max, G. soja | G. latifolia, G. tomentella |
Habitat-Specific Adaptation
Ecological adaptation to diverse habitats represents another significant driver of NLR evolution. Perennial wild relatives of soybean inhabit varied environments across Australia, including deserts, sandy beaches, rocky outcrops, and monsoonal, temperate, and subtropical forests [26]. This ecological diversity creates distinct pathogen pressures that shape species-specific NLR repertoires.
The link between ecological adaptation and NLR evolution extends beyond the Glycine genus. A comprehensive analysis of NLR genes across land plants reveals that NLR gene expansion and contraction are largely driven by ecological adaptation [3]. This pattern is consistent with the concept that plants inhabiting different ecological niches encounter distinct pathogen communities, creating localized selective pressures that shape NLR repertoires through birth-and-death evolution.
Experimental Approaches for Studying NLR Evolution
Genomic Identification and Classification
The identification and classification of NLR genes relies on sophisticated bioinformatic pipelines. The standard methodology involves using PfamScan.pl HMM search script with default e-value (1.1e-50) and the background Pfam-A_hmm model to screen for NBS (NB-NRC) domain-containing genes [19]. All genes containing the NB-ARC domain are considered NBS genes and filtered for further analysis. Additional associated decoy domains are characterized through domain architecture analysis, with genes bearing similar domain architectures placed under the same classes [19].
OrthoFinder v2.5.1 package tools are employed for evolutionary analysis, utilizing the DIAMOND tool for fast sequence similarity searches among NLR sequences [19]. The MCL clustering algorithm facilitates gene clustering, while orthologs and orthogrouping are carried out with DendroBLAST [19]. Multiple sequence alignment is performed using MAFFT 7.0, and gene-based phylogenetic trees are constructed using the maximum likelihood algorithm in FastTreeMP with 1000 bootstrap values [19].
Functional Validation Methods
Functional validation of NLR genes employs several established experimental approaches. Virus-induced gene silencing (VIGS) has been successfully used to demonstrate the functional role of specific NLRs in disease resistance, as shown with GaNBS (OG2) in resistant cotton, where silencing resulted in increased virus titers [19].
Protein-ligand and protein-protein interaction studies provide insights into NLR function, with experiments demonstrating strong interaction between putative NBS proteins and ADP/ATP, as well as with core proteins of the cotton leaf curl disease virus [19]. These interactions are critical for understanding the molecular mechanisms of NLR activation and signaling.
Transcriptomic analyses through RNA-seq data from various databases, including the IPF database, Cotton Functional Genomics Database (CottonFGD), and Cottongen database, enable expression profiling of NLR genes across different tissues and stress conditions [19]. The extracted FPKM values are categorized into biotic stress, abiotic stress, and tissue-specific expression patterns to understand NLR regulation in different contexts.
Diagram 2: Experimental workflow for studying NLR evolution. The methodology progresses from genomic identification through evolutionary analysis to functional validation.
Table 3: Essential Research Reagents for NLR Evolutionary Studies
| Resource Category | Specific Examples | Function/Application |
|---|---|---|
| Genomic Databases | NCBI, Phytozome, Plaza, LegumeInfo | Source of genome assemblies and annotations |
| Bioinformatic Tools | PfamScan.pl, OrthoFinder v2.5.1, DIAMOND, MCL, MAFFT 7.0 | NLR identification, orthogrouping, phylogenetic analysis |
| Expression Databases | IPF Database, CottonFGD, Cottongen | Tissue-specific and stress-responsive expression data |
| Functional Validation Tools | Virus-Induced Gene Silencing (VIGS) | Functional characterization of NLR genes |
| Experimental Resources | Diverse plant accessions (e.g., 17 A. thaliana accessions) | Assessing intraspecific NLR variation |
The evolution of NLR genes in land plants represents a complex interplay between pathogen pressure, ecological adaptation, and genomic constraints. The dramatic expansion of NLR repertoires in flowering plants, coupled with extraordinary structural diversity, underscores the critical role of these genes in plant survival and adaptation. The distinct evolutionary patterns observed between annual and perennial life history strategies further highlight how ecological factors shape immune gene evolution.
Future research in NLR evolution will benefit from several emerging approaches. Pangenome studies across multiple accessions of model and crop species will provide unprecedented resolution of intraspecific NLR diversity [24]. Functional characterization of NLR signaling mechanisms, particularly the co-evolution of sensor and helper NLRs, will reveal how immune signaling networks evolve complexity [3]. Finally, integrating evolutionary studies with crop improvement programs will enable the translation of basic knowledge about NLR diversity into enhanced disease resistance in agricultural systems.
The study of NLR evolution continues to provide fundamental insights into plant-pathogen interactions while offering practical applications for crop improvement. As genomic technologies advance, our understanding of how pathogen pressure and ecological adaptation drive immune gene diversity will continue to deepen, revealing new principles of plant evolution and immunity.
Modern Pipelines for NLR Discovery and Functional Characterization
This technical guide outlines a comprehensive workflow for the genome-wide identification and evolutionary analysis of Nucleotide-binding Leucine-rich repeat (NLR) genes across land plants. NLR genes constitute one of the most dynamic and diverse gene families in plant genomes, serving as critical intracellular immune receptors that mediate effector-triggered immunity. The protocol integrates hidden Markov model (HMMER)-based domain searches with comparative genomic approaches to trace the complex evolutionary history of NLR genes from early land plants like mosses to advanced dicots. We provide detailed methodologies for identification, classification, phylogenetic reconstruction, and evolutionary analysis, along with visual workflows and reagent solutions to facilitate consistent application across research programs. This framework enables researchers to investigate lineage-specific expansions and contractions, gene loss events, and functional diversification of NLR genes throughout plant evolution.
NLR genes encode intracellular immune receptors that recognize pathogen effector proteins and initiate robust defense responses, including the hypersensitive response [27]. These proteins typically contain three fundamental domains: an N-terminal coiled-coil (CC) or toll/interleukin-1 receptor (TIR) domain, a central nucleotide-binding adaptor shared with APAF-1, R proteins, and CED-4 (NB-ARC) domain, and a C-terminal leucine-rich repeat (LRR) domain [27]. Plant NLRs are broadly classified into TNLs (TIR-NB-LRR), CNLs (CC-NB-LRR), and RNLs (RPW8-NB-LRR) based on their N-terminal domains [7].
The NLR gene family exhibits remarkable diversity across land plants, with substantial variation in repertoire size and composition between species. While bryophytes like Physcomitrella patens possess relatively small NLR repertoires (approximately 25 NLRs), angiosperms often contain hundreds of NLR genes [19]. Recent studies have identified 12,820 NBS-domain-containing genes across 34 species covering lineages from mosses to monocots and dicots, revealing both classical and species-specific structural patterns [19]. This diversity results from continuous evolutionary arms races between plants and their pathogens, driving rapid gene duplication, neofunctionalization, and frequent gene loss events [7] [27].
Understanding NLR gene evolution requires specialized bioinformatic approaches that account for their sequence diversity, complex domain architecture, and clustered genomic arrangement. This guide provides detailed protocols for HMMER-based identification and comparative genomic analysis of NLR genes across diverse plant lineages, enabling researchers to reconstruct evolutionary patterns from early land plants to modern angiosperms.
HMMER-Based Workflow for NLR Identification
Domain Identification and Sequence Retrieval
The initial critical step in NLR annotation involves comprehensive identification of candidate sequences containing the conserved NB-ARC domain (Pfam accession: PF00931) using HMMER software suite. The standard workflow proceeds as follows:
Retrieve HMM Profile: Download the NB-ARC (PF00931) hidden Markov model profile from the Pfam database (http://pfam.xfam.org/).
HMMER Search: Execute a domain search against the target proteome using HMMER's
hmmsearchfunction with a conservative E-value threshold (e.g., 10⁻⁴) to maximize sensitivity [7]:Validation with HMMER: Confirm NB-ARC domain presence in candidate sequences using
hmmscanagainst the local Pfam-A database:BLAST Enhancement: Augment HMMER results with a BLASTp search using confirmed NLR sequences as queries (E-value = 1.0) to identify divergent homologs that may have been missed by HMMER [7].
For nucleotide datasets without annotated gene models, NLR-Annotator provides an optimized pipeline that performs six-frame translation prior to HMMER analysis, significantly improving sensitivity for fragmented or unannotated genomes [27].
NLR Classification and Motif Analysis
Following identification, classify NLR genes into subfamilies and characterize conserved motifs:
Subfamily Classification: Differentiate between CNL, TNL, and RNL subclasses through identification of N-terminal domains:
- CNLs: Identify coiled-coil domains using tools like DeepCoil or MARCOIL
- TNLs: Detect TIR domains (Pfam: PF01582) via HMMER search
- RNLs: Recognize RPW8 domains (Pfam: PF05659) in N-terminal regions
Motif Discovery: Identify conserved sequence patterns using MEME Suite with parameters optimized for NLR diversity [27]:
Motif Validation: Assess biological significance of discovered motifs through comparison with known NLR motifs (e.g., P-loop, RNBS-A, MHD) and structural validation.
Table 1: Software Tools for NLR Identification and Annotation
| Tool | Input Type | Methodology | Key Features | Reference |
|---|---|---|---|---|
| NLRtracker | Protein sequences | Integrated InterProScan + custom filters | High sensitivity for full-length NLRs | [27] |
| NLR-Annotator | Nucleotide sequences | Six-frame translation + MAST | Works with unannotated genomes | [28] [27] |
| NLR-Parser | Nucleotide/Protein | MAST output parsing | Biologically curated motif compositions | [28] |
| NLR-Annotator v2.1 | Nucleotide sequences | MEME/MAST pipeline | Command-line implementation | [27] |
Figure 1: HMMER-based workflow for NLR gene identification
Comparative Genomics Framework
Phylogenetic Analysis and Orthogroup Inference
Reconstruct evolutionary relationships among identified NLR genes using phylogenetic approaches:
Sequence Alignment: Extract NB-ARC domain sequences and perform multiple sequence alignment using MAFFT with accuracy-oriented parameters [27]:
Phylogenetic Reconstruction: Construct maximum likelihood trees with model testing:
Orthogroup Delineation: Identify orthologous groups across species using OrthoFinder with default parameters [19]:
Evolutionary Pattern Analysis
Comparative analysis of NLR gene repertoires across land plants reveals distinct evolutionary patterns:
Table 2: Evolutionary Patterns of NLR Genes Across Plant Lineages
| Plant Lineage | Representative Species | NLR Count | Evolutionary Pattern | Key Features | |
|---|---|---|---|---|---|
| Bryophytes | Physcomitrella patens | ~25 | Minimal expansion | Ancestral NLR repertoire | [19] |
| Lycophytes | Selaginella moellendorffii | ~2 | Extreme contraction | Limited NLR diversity | [19] |
| Apiaceae | Coriandrum sativum | 183 | Expansion/Contraction | Species-specific variation | [7] |
| Apiaceae | Angelica sinensis | 95 | Contraction | Significant gene loss | [7] |
| Woody Angiosperms | Eucalyptus grandis | Lineage-specific patterns | Gene loss in specific subgroups | Absence of Group III MDHs | [29] |
| Brassica | Brassica species | First expansion then contraction | Post-WGD evolution | Distinct from Poaceae | [7] |
Application of this framework to Apiaceae species revealed that NLR genes were derived from 183 ancestral NLR lineages and experienced different levels of gene-loss and gain events, with Daucus carota showing contraction while Coriandrum sativum and Apium graveolens exhibited expansion followed by contraction [7].
Genomic Distribution and Synteny Analysis
Characterize genomic organization of NLR genes and identify evolutionary events:
Cluster Identification: Define NLR clusters using sliding-window analysis (250kb window) where intergenic distance < 250kb [7].
Synteny Mapping: Identify conserved syntenic blocks using MCScanX with BLASTP all-against-all results:
Duplication Typing: Classify gene duplication events into WGD, tandem, segmental, or dispersed categories using duplicategeneclassifier in MCScanX.
Figure 2: Comparative genomics workflow for NLR evolutionary analysis
Table 3: Essential Research Reagents and Computational Tools for NLR Genomics
| Category | Resource | Specifications | Application | Source |
|---|---|---|---|---|
| HMM Profiles | NB-ARC (PF00931) | Curated seed alignment | Core domain identification | Pfam Database |
| HMM Profiles | TIR (PF01582) | TIR-domain specific | TNL subclassification | Pfam Database |
| Software | HMMER v3.4 | Command-line tool | Domain searching | http://hmmer.org/ |
| Software | MEME Suite v5.5.5 | Motif discovery | Conserved motif identification | https://meme-suite.org/ |
| Software | NLRtracker v1.0.3 | Integrated pipeline | Automated NLR annotation | [27] |
| Software | OrthoFinder v2.5.1 | Python-based | Orthogroup inference | [19] |
| Software | MCScanX | Java-based | Synteny and duplication analysis | [7] |
| Databases | Pfam v30.0 | HMM database | Domain verification | http://pfam.xfam.org/ |
| Databases | CoGe | Comparative genomics | Genome comparisons | https://genomevolution.org/coge/ |
The integration of HMMER-based domain identification with comparative genomics provides a powerful framework for elucidating the evolutionary history of NLR genes across land plants. This workflow enables researchers to track lineage-specific expansions and contractions, identify key duplication events, and correlate genomic changes with ecological adaptations. As genome sequencing technologies advance and more plant genomes become available, these methodologies will continue to refine our understanding of how NLR repertoires have evolved from simple ancestral forms in early land plants to the complex, diversified arrays found in modern angiosperms. The protocols and resources presented here offer a standardized approach for investigating NLR gene evolution across the plant kingdom, facilitating comparative studies that can reveal fundamental principles of plant-pathogen co-evolution.
The plant immune system, governed primarily by Nucleotide-binding Leucine-rich Repeat (NLR) proteins, represents one of the most dynamic and complex gene families in plant genomes. Recent advances in pangenomics have revolutionized our understanding of NLR evolution, moving beyond single-reference genomes to capture the full spectrum of variation across entire species. This technical guide explores how pangenomic approaches are unraveling the extraordinary intraspecific diversity of NLR genes in Arabidopsis thaliana. By integrating data from multiple genome assemblies, transcriptomic analyses, and epigenomic profiling, researchers are now identifying the evolutionary mechanisms that generate NLR diversity and maintain functional immune systems. These findings provide critical insights into the molecular arms race between plants and pathogens, with implications for breeding disease-resistant crops and understanding fundamental evolutionary processes in land plants from mosses to dicots.
NLR (NOD-like receptor) genes constitute one of the largest and most diverse gene families in plants, encoding intracellular immune receptors that recognize pathogen effector proteins and trigger defense responses [30]. These proteins typically consist of three principal domains: an N-terminal coiled-coil (CC) or toll/interleukin-1 receptor (TIR) domain, a central nucleotide-binding domain (NB-ARC), and a C-terminal leucine-rich repeat (LRR) domain [27]. The NLR family is divided into several subclasses, including TNL (TIR-NB-LRR), CNL (CC-NB-LRR), and RNL (RPW8-NB-LRR), which differ in their structural features and signaling pathways [30].
Traditional genomics, relying on single reference genomes, provided an incomplete picture of NLR diversity due to their remarkable sequence variation and complex genomic organization. Pangenomics—the construction of genomic graphs that represent sequence variation across multiple individuals—has enabled breakthrough discoveries in NLR biology [18]. By analyzing 17 diverse Arabidopsis thaliana accessions, researchers have annotated 3,789 NLRs and defined 121 pangenomic NLR neighborhoods that vary dramatically in size, content, and complexity [18]. This reference-agnostic perspective has revealed that NLR classification based on a single reference genome rarely captures all major paralogs in a cluster accurately [31].
In the broader context of land plant evolution, NLR genes show fascinating patterns of expansion and contraction. Recent studies have revealed that NLR reduction is associated with ecological specialization, particularly in aquatic, parasitic, and carnivorous plants [21]. The convergent NLR reduction in aquatic plants resembles the lack of NLR expansion during the long-term evolution of green algae before the colonization of land, suggesting that the terrestrial environment drove NLR diversification [21].
Patterns of NLR Diversity in Arabidopsis
Classification of NLR Genes: hvNLRs versus non-hvNLRs
Pangenomic studies in Arabidopsis have revealed that NLR genes can be broadly categorized into two distinct types based on their variability patterns:
Table 1: Characteristics of hvNLRs versus non-hvNLRs in Arabidopsis
| Feature | hvNLRs (Highly Variable NLRs) | non-hvNLRs (Low-Variability NLRs) |
|---|---|---|
| Amino acid diversity | High Shannon entropy (>1.5 bits at ≥10 positions) [32] | Low Shannon entropy [32] |
| Selection pressure | Diversifying selection at specific sites [32] | Purifying selection [32] |
| Expression level | Significantly higher [32] | Lower [32] |
| Gene body CG methylation | Significantly lower [32] | Higher [32] |
| Proximity to TEs | Closer to transposable elements [32] | Further from TEs [32] |
| Functional associations | Direct effector recognition [32] | Indirect recognition [32] |
| Cluster organization | Often in radiating clusters [31] | Often as high-fidelity genes [31] |
Genomic Distribution and Cluster Organization
NLR genes in Arabidopsis demonstrate non-random genomic distribution patterns with significant evolutionary implications:
- Cluster-centric expansion: Massive multi-gene NLR cluster expansion does not typically span whole clusters but is restricted to a handful of, or only one, dominant radiations [31].
- Reference-agnostic perspective: Classification of NLRs using gene IDs from a single reference accession (e.g., Columbia-0) rarely captures all major paralogs in a cluster accurately [31].
- Distinct evolutionary processes: Different evolutionary mechanisms act on NLR neighborhoods defending against biotrophic pathogens, with diversity arising from multiple uncorrelated mutational and genomic processes [18].
Table 2: Scale of Pangenomic NLR Studies in Arabidopsis
| Study Aspect | Scale | Key Finding |
|---|---|---|
| Arabidopsis accessions analyzed | 17-64 accessions [18] [31] | Extensive presence-absence variation and structural polymorphisms |
| NLRs annotated | 3,789 NLRs [18] | 121 pangenomic NLR neighborhoods with varying complexity |
| Cluster size variation | Up to 66-fold among closely related species [21] | Rapid gene loss and gain drives diversity |
| Shannon entropy threshold | ≥1.5 bits at ≥10 positions defines hvNLRs [32] | Bimodal distribution of diversity in NLRome |
Methodologies for Pangenomic NLR Analysis
NLR Annotation and Identification
The accurate annotation of NLR genes from genomic sequences requires specialized computational tools and pipelines:
Figure 1: Workflow for NLR Gene Annotation
Step-by-Step Protocol for NLR Annotation:
Data Acquisition: Download protein or nucleotide sequences from reference genome databases. For proteome-wide analysis, compile sequences into a single FASTA file [27].
Tool Selection: Choose appropriate annotation software based on data type:
Execution Command:
The output NLR protein sequences are typically saved as "NLR.fasta" in the specified output directory [27].
Validation: Cross-reference annotations with known functionally characterized NLRs to ensure completeness, as some tools may miss validated NLRs like ADR1 [27].
Phylogenomic Analysis and Motif Discovery
Following annotation, phylogenetic analysis enables classification of NLRs into subfamilies and identification of conserved motifs:
Procedure for Phylogenomic Analysis:
Sequence Alignment: Use MAFFT v7 for multiple sequence alignment of identified NLR sequences [27].
Phylogenetic Tree Construction: Employ RAxML v8.2.12 for maximum likelihood-based inference of large phylogenetic trees [27].
Subfamily Extraction: Classify NLRs based on phylogenetic clustering and extract specific subfamily sequences (e.g., CC-NLRs) for further analysis [27].
Motif Discovery: Utilize MEME Suite v5.5.5 for identifying conserved sequence motifs within NLR subfamilies:
This approach has successfully identified functionally important motifs like MADA and EDVID in CC-NLR subfamilies [27].
Pangenome Graph Construction
The core innovation enabling comprehensive NLR analysis is pangenome graph construction:
Data Integration: Combine genome assemblies from multiple accessions with full-length transcript data, homology information, and transposable element annotations [18].
Graph Construction: Build pangenome graphs that capture sequence variations, presence-absence polymorphisms, and structural variations across accessions.
Neighborhood Definition: Define NLR neighborhoods based on synteny and sequence similarity, allowing for comparative analysis of cluster organization and complexity [18].
Evolutionary Mechanisms Generating NLR Diversity
The extensive intraspecific variation in NLR genes arises through multiple evolutionary mechanisms:
Figure 2: Evolutionary Mechanisms Driving NLR Diversity
Genetic Processes
Unequal crossing over and gene conversion: These processes drive NLR expansion and diversification in clusters, which occur more frequently for NLRs than other genes [32].
Point mutations: A major source of within-species NLR diversity, resulting in the most polymorphic loci in the Arabidopsis genome with the highest frequency of major effect mutations [32].
Transposable element association: hvNLRs are significantly closer to transposable elements, which can promote rapid diversification through various mutagenic mechanisms [32].
Selection Pressures
Balancing selection: Maintains polymorphisms through frequency-dependent selection, spatial and temporal fluctuations in pathogen pressure, and heterozygote advantage [32].
Diversifying selection: Observed as an excess of nonsynonymous to synonymous substitutions at specific codons, particularly in hvNLRs [32].
Purifying selection: Maintains conservation in non-hvNLRs, preserving essential functions in immune signaling [32].
Research Reagents and Computational Tools
Table 3: Essential Research Reagents and Tools for Pangenomic NLR Studies
| Tool/Reagent | Type | Function | Application in NLR Research |
|---|---|---|---|
| NLRtracker | Software | NLR annotation from protein sequences | Identifies NLR genes from proteome datasets [27] |
| NLR-Annotator | Software | NLR annotation from nucleotide sequences | Alternative for users without Linux systems [27] |
| InterProScan | Software | Protein function characterization | Domain analysis in NLRtracker pipeline [27] |
| MAFFT | Software | Multiple sequence alignment | Aligns NLR sequences for phylogenetic analysis [27] |
| RAxML | Software | Phylogenetic tree inference | Constructs evolutionary relationships of NLRs [27] |
| MEME Suite | Software | Motif-based sequence analysis | Discovers conserved sequence patterns in NLRs [27] |
| MCL | Software | Clustering weighted networks | Groups NLRs into protein families [27] |
| BLAST+ | Software | Sequence similarity search | Identifies homologous NLR sequences [27] |
| HMMER | Software | Sequence homology search | Finds sequence homologs in databases [27] |
| Long-read sequencing | Technology | Genome assembly | Resolves complex NLR regions [31] |
Discussion and Future Perspectives
Pangenomic approaches have fundamentally transformed our understanding of NLR evolution in Arabidopsis and other plant species. The integration of pangenome graphs with epigenomic data has revealed how genomic features like gene body methylation and TE proximity create mutation biases that shape NLR diversity [32]. These findings have profound implications for understanding plant-pathogen coevolution and developing sustainable crop protection strategies.
The distinction between hvNLRs and non-hvNLRs provides a framework for understanding how plants balance the need for innovative recognition specificities against the metabolic costs of maintaining a large, diverse immune receptor repertoire [30] [32]. The association between hvNLRs and specific genomic features suggests mechanistic links between mutation rate variation and selection pressures.
Future research directions should include:
- Mechanistic studies: Determining how epigenomic features directly influence mutation rates in NLR genes.
- Functional validation: Testing the recognition specificities of diverse NLR alleles against pathogen effectors.
- Broad taxonomic sampling: Applying pangenomic approaches across the land plant phylogeny to understand NLR evolution from mosses to dicots.
- Breeding applications: Leveraging natural NLR diversity to develop durable disease resistance in crops.
The pangenomic perspective has revealed that "diversity in diversity generation" is fundamental to maintaining a functionally adaptive immune system in plants [18]. This insight, coupled with the methodological advances described in this guide, provides a powerful framework for unraveling the evolutionary dynamics of plant immunity.
In the study of plant immunity, the evolution of nucleotide-binding leucine-rich repeat (NLR) genes represents a central adaptive mechanism in the enduring conflict between plants and their pathogens. These genes, which encode intracellular receptors responsible for detecting pathogen effectors and initiating robust defense responses, have undergone remarkable expansion and diversification throughout plant evolutionary history [1]. From the relatively simple NLR repertoires of non-vascular mosses to the highly complex and variable collections found in dicots, understanding which specific NLR genes merit in-depth functional characterization presents a significant research challenge. This whitepaper details a comprehensive methodology that leverages transcriptomics data to prioritize NLR candidate genes by treating gene expression patterns as functional signatures indicative of biological importance. We frame this approach within a comparative evolutionary context, tracing NLR development from early land plants like mosses to advanced dicot species.
Evolutionary Context of NLR Genes in Land Plants
NLR proteins are fundamental components of the plant innate immune system, characterized by a conserved domain architecture typically consisting of a central nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4 (NB-ARC) domain, a C-terminal leucine-rich repeat (LRR) domain, and an N-terminal toll/interleukin-1 receptor (TIR) or coiled-coil (CC) domain [1] [27]. The evolutionary trajectory of these genes reveals a story of extensive diversification and expansion correlated with increasing plant complexity.
NLR Repertoire Expansion Across Plant Lineages
Comparative genomics analyses reveal dramatic differences in NLR repertoire sizes across the plant kingdom, as detailed in Table 1. Early divergent land plant lineages, such as the moss Physcomitrella patens, possess relatively small NLR repertoires of approximately 25 genes, while flowering plants exhibit substantial expansions, with some species containing over 450 NLR genes [1]. This expansion reflects the continuous evolutionary arms race between plants and their fast-evolving pathogens.
Table 1: NLR Gene Repertoire Size Across Representative Plant Species
| Species | Common Name | NLRs | TNLs | CNLs | XNLs | Reference |
|---|---|---|---|---|---|---|
| Physcomitrella patens | Moss | ~25 | 8 | 9 | 8 | [1] |
| Selaginella moellendorffii | Spike moss | ~2 | 0 | NA | NA | [1] |
| Arabidopsis thaliana | Thale cress | 151 | 94 | 55 | 0 | [1] |
| Oryza sativa | Rice | 458 | 0 | 274 | 182 | [1] |
| Vitis vinifera | Wine grape | 459 | 97 | 215 | 147 | [1] |
Mosses as Models for Ancient Immune Mechanisms
Mosses, particularly Physcomitrella patens, occupy a crucial evolutionary position as early divergent land plants and serve as valuable models for understanding the ancestral state of plant immunity [33]. They exhibit resistance to many pathogens that affect vascular plants and possess conserved NLR-mediated defense mechanisms. Research on moss-pathogen interactions with organisms like Botrytis cinerea and Pseudomonas syringae has demonstrated that fundamental components of NLR signaling were established early in land plant evolution [33]. Studying these basal species helps identify evolutionarily conserved immune mechanisms that have been maintained throughout plant evolution, making them excellent starting points for identifying functionally important NLR candidates.
Transcriptomics-Based Candidate Prioritization Framework
The core premise of our approach is that genes with similar expression patterns across evolutionary lineages, in response to specific stimuli, or in particular tissues may share functional similarities and biological importance. For NLR genes, this translates to prioritizing those that show conserved expression signatures associated with defense responses.
Workflow for Transcriptomics-Based NLR Prioritization
The following diagram illustrates the comprehensive workflow for prioritizing NLR candidate genes using transcriptomics data within an evolutionary framework:
Key Methodological Components
NLR Gene Annotation
Accurate identification of NLR genes from genomic or transcriptomic data is the foundational step. Specialized tools have been developed for this purpose:
- NLRtracker: Utilizes protein sequence files as input and integrates InterProScan for domain characterization to identify NLRs with high sensitivity and accuracy [27].
- NLR-Annotator: Works with nucleotide sequence files and is suitable for users without access to Linux systems [27].
For a typical analysis across multiple species, proteomes are downloaded from reference genome databases and compiled into a single FASTA file for annotation. In a representative analysis of six plant species (Arabidopsis thaliana, Beta vulgaris, Solanum lycopersicum, Nicotiana benthamiana, Oryza sativa, and Hordeum vulgare), this approach identified 1,862 NLRs from compiled protein sequences [27].
Transcriptomics Data Analysis
RNA-sequencing technology enables genome-wide expression profiling to identify differentially expressed genes under various conditions. Key considerations include:
- Experimental Design: Ensure sufficient statistical power through appropriate sample sizes and replication to detect true differential expression [34].
- Differential Expression Analysis: Compare transcript abundance between conditions (e.g., pathogen-infected vs. mock-treated tissues) using statistical frameworks that control false discovery rates (FDR) [34].
- Cross-Species Expression Conservation: Identify NLR genes with conserved expression patterns across evolutionary lineages, particularly those consistently upregulated in response to pathogen challenge.
Evolutionary Conservation Assessment
The prioritization of candidate genes can be enhanced by incorporating evolutionary conservation metrics [34]. Genes with sequences and expression patterns conserved across species are more likely to perform essential biological functions. For NLR genes, this involves:
- Phylogenetic Analysis: Construct maximum likelihood phylogenetic trees using tools like RAxML to classify NLRs into subfamilies and identify orthologous genes across species [27].
- Conserved Motif Discovery: Use motif-based sequence analysis tools like MEME Suite to identify functionally important sequence patterns that have been maintained through evolution [27].
Multi-Criteria Scoring System
A systematic scoring approach evaluates candidate NLR genes across multiple biologically relevant criteria. We propose a weighted scoring system (0-10 points per category) based on:
- Expression Responsiveness: Magnitude and significance of differential expression in response to immune challenges.
- Evolutionary Conservation: Sequence and expression pattern conservation across species from mosses to dicots.
- Functional Evidence: Prior genetic or biochemical evidence of immune function.
- Pathway Association: Membership in co-expressed gene modules or known immune signaling pathways.
- Pangenomic Context: Presence and variation across pangenome analyses of multiple accessions [24] [18].
Integrated Protocol for NLR Phylogenomics and Motif Discovery
To identify functionally important NLR candidates, we present a detailed protocol combining phylogenomics and conserved motif discovery, adapted from established methodologies [27].
Phylogenomics Workflow for Conserved Motif Identification
The following diagram outlines the specific workflow for identifying evolutionarily conserved motifs in NLR proteins through phylogenomics:
Step-by-Step Protocol
Step 1: Data Acquisition and NLR Annotation
- Download proteomes from reference genome databases for species of interest (e.g., Arabidopsis, tomato, rice, barley, mosses).
- Compile protein sequences into a single FASTA file.
- Annotate NLRs using NLRtracker:
./NLRtracker -s input_protein.fasta -o output_directory[27]. - Combine identified NLR sequences with known functionally characterized NLRs for reference.
Step 2: Phylogenetic Analysis
- Perform multiple sequence alignment using MAFFT.
- Construct maximum likelihood phylogenetic trees using RAxML.
- Visualize and manipulate trees using iTOL (Interactive Tree Of Life).
- Classify NLRs into subfamilies (TNL, CNL, etc.) based on phylogenetic relationships.
Step 3: Subfamily-Specific Motif Discovery
- Extract sequences for specific NLR subfamilies of interest (e.g., CC-NLRs).
- Use MEME Suite to identify conserved sequence motifs within each subfamily.
- Validate motif conservation across evolutionary lineages from mosses to dicots.
- Characterize newly identified motifs through comparison with known functional motifs (e.g., P-loop, MHD, MADA, EDVID).
Step 4: Integration with Transcriptomics Data
- Map expression data to phylogenetic clusters to identify subfamilies with coordinated expression patterns.
- Prioritize NLR candidates showing both expression responsiveness and evolutionary conservation of key functional motifs.
- Validate selected candidates through site-directed mutagenesis of identified motifs and functional assays in model systems.
Research Reagent Solutions
The following table provides essential research reagents and computational tools for implementing the described NLR prioritization and characterization pipeline.
Table 2: Essential Research Reagents and Tools for NLR Transcriptomics
| Category | Tool/Reagent | Specific Function | Application Notes |
|---|---|---|---|
| NLR Annotation | NLRtracker v1.0.3 | Annotation of NLR genes from protein sequences | Higher sensitivity for detecting diverse NLRs; requires Linux OS [27] |
| NLR Annotation | NLR-Annotator v2.1 | Annotation of NLR genes from nucleotide sequences | Suitable for non-Linux users; works with nucleotide data [27] |
| Sequence Alignment | MAFFT v7 | Multiple sequence alignment | Handles large datasets efficiently [27] |
| Phylogenetics | RAxML v8.2.12 | Maximum likelihood phylogenetic inference | Robust for large phylogenetic trees [27] |
| Motif Discovery | MEME Suite v5.5.5 | Identification of conserved sequence motifs | Web-based or local installation; identifies ungapped motifs [27] |
| Expression Analysis | R/Bioconductor | Differential expression analysis | DESeq2, edgeR for statistical analysis of RNA-seq data [34] |
| Network Analysis | MCL v14-137 | Clustering of co-expressed genes | Identifies functional modules from expression networks [27] |
| Homology Search | BLAST+ v2.12.0 | Identification of homologous sequences | Finds evolutionary relatives across species [27] |
The integration of transcriptomics with evolutionary analysis provides a powerful framework for prioritizing NLR candidate genes in plant immunity research. By treating expression patterns as functional signatures and tracing these patterns across the evolutionary continuum from mosses to dicots, researchers can identify the most biologically relevant NLR genes for in-depth functional characterization. The protocols and workflows detailed in this technical guide offer a systematic approach for leveraging the vast amounts of available genomic and transcriptomic data to advance our understanding of plant immune system evolution and function. This strategy not only accelerates the pace of discovery but also ensures that research efforts are focused on evolutionarily conserved mechanisms with greatest potential for informing crop improvement strategies.
High-throughput functional validation represents a paradigm shift in plant innate immunity research, enabling the systematic discovery of nucleotide-binding domain and leucine-rich repeat receptors (NLRs) at an unprecedented scale. This technical guide details an integrated pipeline that leverages transcriptional signatures, transgenic arrays, and large-scale phenotyping to identify functional NLR immune receptors across diverse plant species. The methodology demonstrates particular utility for characterizing the evolutionary conservation and diversification of NLR genes from basal land plants like mosses to advanced dicot species, providing crucial insights into the molecular architecture of plant immunity through deep evolutionary time.
Evolutionary Context of NLR Genes in Land Plants
NLR immune receptors constitute one of the most dynamically evolving gene families in plant genomes, exhibiting remarkable structural and functional diversification throughout plant evolutionary history. From the basal bryophytes (including mosses) to tracheophytes comprising ferns, gymnosperms, and angiosperms (both monocots and dicots), NLRs have expanded via repeated duplication events and functional specialization. This evolutionary trajectory reflects an ongoing molecular arms race with rapidly evolving pathogens across different plant lineages. The conservation of NLR signaling mechanisms from mosses to dicots suggests ancient origins for core immune signaling components, while lineage-specific expansions reveal adaptive innovations to particular pathogen pressures. Understanding this evolutionary continuum requires functional validation tools capable of operating across phylogenetic boundaries, which high-throughput validation pipelines now provide.
Core Experimental Workflow and Methodologies
Expression-Based Candidate Prioritization
The foundational principle of this approach recognizes that functional NLRs often exhibit characteristic high expression signatures in uninfected plants across monocot and dicot species [35]. This expression pattern facilitates bioinformatic prioritization from genomic or transcriptomic datasets.
Protocol: Expression Signature Analysis
- Data Collection: Compile RNA-seq datasets from uninfected leaf tissues across target species (from mosses to dicots)
- NLR Identification: Annotate NLR complement using domain-based search tools (NB-ARC, LRR domains)
- Expression Quantification: Calculate transcripts per million (TPM) values for all NLR genes
- Candidate Selection: Prioritize NLRs falling within the top 15% of expressed NLR transcripts, as this segment shows significant enrichment for functionally validated receptors (χ² test, P = 0.038) [35]
- Cross-Species Comparison: Identify orthologous NLR groups across evolutionary lineages to trace conservation patterns
High-Throughput Transformation Array Construction
A transgenic array platform enables parallel functional testing of hundreds to thousands of NLR candidates in a uniform genetic background.
Protocol: Wheat Transformation Array [35]
- Vector Construction: Clone 995 NLR candidate genes from diverse grass species into binary transformation vectors under appropriate regulatory sequences (native promoters or constitutive promoters like Ubiquitin)
- Plant Material Preparation: Use immature embryos of susceptible wheat cultivars as explant tissue
- Transformation Method: Employ Agrobacterium tumefaciens-mediated transformation with modified protocols for high efficiency [35]
- Selection and Regeneration: Apply appropriate antibiotic/herbicide selection followed by plant regeneration on hormone-containing media
- Transgene Verification: Confirm integration via PCR and copy number estimation using digital PCR or Southern blotting
- Line Propagation: Advance transgenic events to T1/T2 generations to ensure stability and homozygosity
Table 1: High-Throughput Transformation Specifications
| Parameter | Specification | Throughput | Efficiency |
|---|---|---|---|
| NLR Candidates | 995 genes from diverse grasses | 995 constructs | N/A |
| Vector System | Binary vectors with selection markers | Standard cloning | >90% cloning success |
| Transformation Method | Agrobacterium-mediated | 100-200 embryos/week | 5-25% transformation efficiency |
| Generation Time | T0 to T2 generations | 8-12 months | N/A |
| Validation Methods | PCR, Southern blot, expression analysis | 50-100 lines/week | >95% verification rate |
Large-Scale Phenotyping for Disease Resistance
Comprehensive pathogen challenge assays identify NLRs conferring resistance against major agricultural pathogens.
Protocol: Rust Disease Phenotyping [35]
- Pathogen Preparation: Maintain and propagate virulent isolates of Puccinia graminis f. sp. tritici (Pgt, stem rust) and Puccinia triticina (Pt, leaf rust)
- Inoculation Method: Apply urediospores suspended in lightweight mineral oil to 10-14 day old seedling leaves using standardized inoculation towers
- Disease Development: Maintain inoculated plants in dew chambers at 18-22°C for 24 hours, then transfer to greenhouse conditions (20-25°C)
- Phenotype Scoring: Assess disease symptoms 12-14 days post-inoculation using standardized rust scoring scales (0-4 for infection type)
- Digital Phenotyping Enhancement: Implement image-based analysis for objective quantification of disease symptoms [36]
- Statistical Analysis: Apply appropriate statistical models to distinguish significant resistance from background variation
Table 2: Phenotyping Outcomes from NLR Transgenic Array
| Pathogen System | NLRs Tested | Functional NLRs Identified | Resistance Efficacy | Evolutionary Origin |
|---|---|---|---|---|
| Stem Rust (Pgt) | 995 | 19 | Race-specific resistance | Diverse grass species |
| Leaf Rust (Pt) | 995 | 12 | Race-specific resistance | Diverse grass species |
| Validation Rate | 995 | 31 (3.1%) | Confirmed function | Cross-genus transfer |
Signaling Pathways and Experimental Workflow
Diagram 1: High-throughput NLR validation workflow.
Diagram 2: NLR-mediated immune signaling pathway.
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for NLR Validation Pipeline
| Reagent/Category | Function/Purpose | Specific Examples/Applications |
|---|---|---|
| NLR Candidate Libraries | Source of diverse immune receptors for testing | 995 NLR genes from diverse grass species; ortholog sets from mosses to dicots |
| Binary Vector Systems | Plant transformation and transgene expression | Agrobacterium-compatible vectors with plant selection markers (hygromycin, basta) |
| Plant Transformation Lines | High-efficiency transformation recipients | Wheat cultivar 'Fielder' with high transformation efficiency [35] |
| Pathogen Isolates | Disease resistance phenotyping | Puccinia graminis f. sp. tritici (stem rust), Puccinia triticina (leaf rust) |
| Phenotyping Platforms | Automated disease scoring and data collection | Digital imaging systems with image analysis algorithms for objective symptom quantification [36] |
| Bioinformatics Tools | NLR identification, expression analysis, evolutionary tracking | Domain annotation pipelines, expression quantification, phylogenetic analysis software |
Discussion and Evolutionary Implications
The integration of high-throughput validation platforms with evolutionary analysis provides unprecedented resolution into NLR gene family dynamics across land plants. Several key insights emerge from this approach:
Evolutionary Conservation and Divergence: The pipeline demonstrates that NLRs maintaining high expression across species boundaries often retain conserved immune functions, suggesting preservation of core signaling mechanisms from ancestral lineages. However, lineage-specific expansions in dicots compared to mosses reveal substantial functional diversification.
Expression-Level Evolution: The correlation between high expression and NLR functionality appears conserved across bryophytes and tracheophytes, indicating ancient regulatory constraints on immune receptor efficacy. This challenges previous assumptions about NLR transcriptional repression and suggests expression level represents an evolutionarily significant parameter.
Cross-Species Transferability: Successful resistance conferral by grass NLRs in wheat demonstrates the functional conservation of immune signaling components across millions of years of evolutionary divergence, highlighting the potential for mining NLRs from basal species for crop protection.
This high-throughput validation framework establishes a powerful platform for interrogating NLR gene evolution across the plant kingdom, from the most basal moss lineages to advanced flowering plants, accelerating both fundamental understanding of plant immunity and applied resistance breeding.
Plant immunity relies on a sophisticated innate immune system where intracellular nucleotide-binding leucine-rich repeat receptors (NLRs) serve as critical components for pathogen recognition and defense activation. These receptors detect pathogen-secreted effector proteins, initiating a robust defense response known as effector-triggered immunity (ETI), often characterized by a hypersensitive response involving programmed cell death at infection sites [37]. NLR genes represent one of the most diverse and rapidly evolving gene families in plants, reflecting the continuous evolutionary arms race between plants and their pathogens [37]. This genetic diversity, particularly in wild relatives of modern crops, contains untapped potential for breeding disease-resistant cultivars. The evolutionary journey of NLR genes from early land plants like mosses to dicots reveals a story of tremendous genetic innovation, with lineage-specific expansions, contractions, and diversification driven by tandem duplication events, domain shuffling, and positive selection [37]. Mining this diversity through advanced bioinformatics approaches, functional characterization, and strategic breeding represents a promising pathway to enhancing crop resilience in the face of evolving pathogen threats.
Evolutionary Perspectives on NLR Diversity Across Plant Lineages
Genomic Architecture and Expansion Patterns
NLR genes exhibit remarkable variation across the plant kingdom, with significant differences in number, composition, and architecture between species. Comparative genomic analyses reveal that NLR content varies dramatically, ranging from approximately 50 genes in species like watermelon (Citrullus lanatus) and papaya (Carica papaya) to over 1,000 in apple (Malus domestica) and hexaploid wheat (Triticum aestivum) [37]. This variation stems from lineage-specific expansions and contractions, predominantly through tandem duplication and deletion events influenced by transposon content, ecological context, and adaptation to local pathogen pressures [37]. Wild plant relatives often harbor greater NLR diversity compared to domesticated crops, as agricultural selection bottlenecks have reduced genetic variation, including at NLR loci.
Table 1: NLR Gene Repertoire Across Selected Plant Species
| Plant Species | Classification | NLR Count | Key Features | Reference |
|---|---|---|---|---|
| Arabidopsis thaliana | Dicot (model) | ~50-200 (species-wide) | High intraspecific diversity; presence-absence variation | [37] |
| Hordeum vulgare (barley) | Monocot | Part of 1,862 NLR dataset | Co-evolved with monocot-specific pathogens | [27] |
| Oryza sativa (rice) | Monocot | Part of 1,862 NLR dataset | Well-characterized NLRs with known resistance specificities | [27] |
| Malus domestica (apple) | Dicot (tree) | >1,000 | Extensive duplication and diversification | [37] |
| Triticum aestivum (wheat) | Monocot (hexaploid) | >1,000 | Complex repertoire due to polyploidy | [37] |
| Dioscorea zingiberensis (yam) | Monocot | Significant expansion | 33.8%-127.5% more NLRs found via reannotation | [38] |
Structural and Functional Diversification Through Evolutionary Time
Plant NLRs share a conserved tripartite domain architecture consisting of an N-terminal domain, a central nucleotide-binding domain (NB-ARC), and C-terminal leucine-rich repeats (LRRs) [37]. The N-terminal domains have diversified into several major classes throughout plant evolution: coiled-coil (CC)-type, RPW8-type (CCR), G10-type CC (CCG10), and toll/interleukin-1 receptor-type (TIR) domains [37]. Non-flowering plants possess additional N-terminal domain types, such as α/β hydrolases and kinase domains, revealing deeper evolutionary origins and functional diversification [37]. While most NLRs retain this canonical structure, many have evolved specialized configurations with additional noncanonical domains or degenerated features, enabling functional specialization within immune networks.
The evolutionary transition from singleton NLR operation to complex pairs and networks represents a key adaptation in plant immunity. In NLR pairs and networks, sensor NLRs specialize in pathogen recognition while helper NLRs transduce immune signals, creating sophisticated many-to-one and one-to-many functional connections that enhance robustness and evolvability [37]. This modular organization allows plants to recognize numerous pathogens with limited genetic resources while maintaining signaling efficiency. Understanding these evolutionary patterns provides the foundation for strategic mining of NLR diversity from wild relatives to enhance cultivated crops.
Technical Approaches for Mining NLR Diversity
Advanced NLR Annotation Pipelines and Tools
Conventional genome annotation pipelines frequently misannotate NLR genes due to their unusual gene structure, sequence diversity, and frequent location in complex genomic regions. To address these challenges, specialized bioinformatics tools have been developed for accurate NLR identification:
NLRtracker: A sensitive annotation tool that uses protein sequence files as input, demonstrating higher accuracy in extracting NLRs from plant proteomes compared to conventional methods [27]. It successfully identified 1,862 NLRs from six representative monocot and dicot species in a test dataset [27].
NLR-Annotator: Suitable for users without Linux systems, this tool works with nucleotide sequence files as input datasets [27].
NLRSeek: A recently developed reannotation-based pipeline that integrates de novo NLR detection at the genome level with targeted reannotation, systematically reconciling results with existing annotations [38]. This approach identified 33.8%-127.5% more NLR genes in yam species compared to conventional methods, with 45.1% of newly annotated NLRs showing detectable expression, confirming they represent functional genes [38].
Table 2: Bioinformatics Tools for NLR Identification and Annotation
| Tool Name | Input Type | Key Features | Advantages | Application Example |
|---|---|---|---|---|
| NLRtracker | Protein sequences | High sensitivity and accuracy | Detects functionally validated NLRs missed by other tools | Identified 1,862 NLRs from 6 plant species [27] |
| NLR-Annotator | Nucleotide sequences | User-friendly, no Linux required | Suitable for diverse computational environments | Alternative to NLRtracker for nucleotide data [27] |
| NLRSeek | Genome sequences | Reannotation-based pipeline | Identifies previously missed NLRs in existing annotations | Found 127.5% more NLRs in yam species [38] |
| InterProScan | Protein sequences | Integrated signature database | Combines multiple motif databases with manual curation | Functional domain characterization [27] |
Phylogenomics and Conserved Motif Identification
Due to the sequence diversity of NLRs, identifying functionally important regions requires specialized phylogenomic approaches that overcome limitations of conventional multiple sequence alignment. A comprehensive computational pipeline has been developed for identifying evolutionarily conserved motifs in plant NLR proteins through the following workflow:
NLR Annotation: Annotate NLRs from proteome datasets using specialized tools (NLRtracker or NLR-Annotator) [27].
Dataset Compilation: Combine annotated NLRs with functionally characterized reference NLRs to create a comprehensive dataset for analysis [27].
Phylogenetic Analysis: Classify NLRs into subfamilies using maximum likelihood-based inference tools (RAxML) following multiple sequence alignment (MAFFT) [27].
Sequence Clustering: Group sequences into protein families using Markov Cluster algorithm (MCL) based on sequence similarity [27].
Motif Discovery: Identify conserved sequence motifs within clusters using motif-based sequence analysis tools (MEME Suite) [27].
This pipeline has successfully identified functionally important motifs like the MADA and EDVID motifs within the CC-NLR subfamily, molecular signatures that have remained conserved throughout evolutionary time across plant species [27]. These conserved motifs represent critical functional domains whose manipulation can enhance NLR function in crop breeding.
Experimental Validation of NLR Function
Computational predictions require experimental validation to confirm NLR function and resistance specificity. Several key methodologies enable this critical step:
Heterologous Expression: Transfer of candidate NLR genes from wild relatives into susceptible crop cultivars to test for resistance complementation [39].
Site-Directed Mutagenesis: Targeted mutations in conserved regions (e.g., P-loop and MHD motifs) to confirm their necessity for NLR function [27].
Transcriptional Profiling: Analysis of gene expression patterns using RNA-Seq to confirm NLR expression and identify differentially expressed genes following pathogen challenge [38].
Ribosome Profiling: Experimental confirmation of translation for newly identified NLR genes validated through reannotation pipelines [38].
NLR Engineering and Breeding Applications
Engineering Autoactive NLRs for Broad-Spectrum Resistance
Recent advances in understanding NLR structure and function have enabled knowledge-guided engineering approaches. A breakthrough strategy involves remodeling autoactive NLRs (aNLRs) for broad-spectrum disease resistance [39]. This approach utilizes chimeric aNLRs comprising an autoactive CNL/RNL (aCNL/aRNL) and an N-terminal blocking peptide coupled with a pathogen-originated protease cleavage site (PCS) [39]. Under normal conditions, the autoactivity is suppressed, but upon infection, pathogen-encoded proteases cleave the PCS, removing the blocking peptide and activating the aNLR to trigger a potent immune response [39].
This innovative strategy offers several advantages over conventional NLR engineering:
- Simpler Design: Requires only a single autoactive RNL or CNL intolerant to N-terminal fusions and a short blocking peptide with PCS [39].
- Broad Recognition Spectrum: Activation depends on cleavage by pathogen-encoded proteases rather than specific effector binding, enabling resistance to multiple pathogens using conserved PCSs [39].
- Durability: Mutations in protease that prevent PCS cleavage would simultaneously compromise pathogen fitness, making resistance more durable [39].
- Cross-Species Applicability: CNLs and RNLs are prevalent in crops, and their calcium channel activity upon resistosome formation is conserved, enabling broad application across diverse crop species [39].
Integrating Wild NLR Diversity into Breeding Programs
Effective utilization of wild NLR diversity in crop breeding requires strategic approaches:
Introgression Breeding: Traditional crossing of crop varieties with wild relatives followed by backcrossing and selection for desired NLR genes alongside agronomic traits.
Marker-Assisted Selection: Development of molecular markers tightly linked to valuable NLR genes for efficient tracking during breeding.
Gene Stacking: Pyramiding multiple NLR genes with complementary resistance spectra into elite cultivars to enhance durability.
Genome Editing: Precise modification of endogenous NLR genes using CRISPR/Cas technology to enhance function or alter specificity [39].
Table 3: Research Reagent Solutions for NLR Mining and Characterization
| Category | Specific Tool/Resource | Function/Application | Key Features |
|---|---|---|---|
| Bioinformatics Tools | NLRtracker | NLR identification from proteome data | High sensitivity, detects functionally validated NLRs [27] |
| NLR-Annotator | NLR annotation from nucleotide sequences | User-friendly, no Linux requirement [27] | |
| NLRSeek | Genome reannotation for missed NLRs | Identifies previously unannotated functional NLRs [38] | |
| MEME Suite | Conserved motif discovery | Identifies evolutionarily conserved sequence patterns [27] | |
| Experimental Resources | RefPlantNLR | Curated collection of validated NLRs | Reference dataset for phylogenetic analysis [37] |
| EggNOG-mapper | Functional annotation transfer | Assigns Gene Ontology terms, KEGG pathways [40] | |
| InterProScan | Protein domain characterization | Integrated database of protein families and domains [40] | |
| Characterization Methods | Site-directed mutagenesis | Functional validation of conserved motifs | Tests necessity of specific residues/domains [27] |
| Ribosome profiling | Translation confirmation | Validates expression of newly identified NLRs [38] | |
| Heterologous expression | Functional testing in model systems | Confirms resistance capability of candidate NLRs [39] |
Mining the untapped NLR diversity present in wild plant relatives offers tremendous potential for enhancing disease resistance in cultivated crops. The evolutionary journey of NLR genes from early land plants to modern dicots has generated a rich reservoir of genetic variation that can be harnessed through integrated approaches combining advanced bioinformatics, phylogenomics, and strategic engineering. The development of specialized tools like NLRtracker and NLRSeek has dramatically improved our ability to identify the complete NLR repertoire in plant genomes, revealing previously overlooked functional genes. Meanwhile, innovative engineering strategies such as autoactive NLR remodeling provide powerful methods to translate this genetic diversity into durable, broad-spectrum resistance. As genomic technologies continue to advance and our understanding of NLR structure-function relationships deepens, the strategic mining of NLR diversity from wild relatives will play an increasingly vital role in developing resilient crop varieties capable of withstanding evolving pathogen threats in changing agricultural environments.
Navigating the Challenges of NLR Regulation and Deployment
The evolution of land plants from simple bryophytes to complex dicots has been shaped by a constant arms race against pathogens. Central to this process is the nucleotide-binding domain and leucine-rich repeat receptor (NLR) family, which constitutes one of the largest and most variable gene families in plant genomes [1]. These intracellular immune receptors recognize pathogen effectors and initiate effector-triggered immunity (ETI), often culminating in a hypersensitive response characterized by programmed cell death at infection sites [30]. While this immune system provides crucial protection against diverse pathogens, it imposes significant metabolic costs that must be carefully balanced against plant growth, development, and reproductive fitness [30] [41]. This conundrum—how plants maintain effective immunity without debilitating self-damage—has driven the evolution of sophisticated regulatory mechanisms that operate across genetic, transcriptional, and protein levels.
The NLR family has undergone massive expansions in flowering plants, with genomes encoding from fewer than 100 to over 2,000 NLR genes, in contrast to the relatively small repertoires of approximately 25 NLRs in the bryophyte Physcomitrella patens and only 2 in the lycophyte Selaginella moellendorffii [1] [2]. This dramatic expansion reflects continuous adaptation to evolving pathogen pressures, yet it also increases the risk of autoimmunity—a state where immune receptors mistakenly recognize self-molecules or become dysregulated, triggering deleterious defense responses in the absence of pathogens [30] [41]. Understanding how plants navigate this delicate balance requires integrated analysis of NLR evolutionary history, molecular regulation, and the fitness consequences of immune activation.
NLR Gene Evolution: Expansion and Diversification Across Land Plants
Comparative Genomic Analysis of NLR Repertoires
The NLR family exhibits remarkable diversity across plant taxa, with significant variation in both the number and composition of NLR genes. Table 1 summarizes the genomic distribution of NLRs across representative plant species, illustrating the patterns of expansion from early land plants to flowering plants.
Table 1: NLR Repertoire Expansion Across Plant Lineages
| Species | Common Name | Plant Group | Genome Size (Mbp) | Total NLRs | TNLs | CNLs | Other NLRs |
|---|---|---|---|---|---|---|---|
| Physcomitrella patens | Moss | Bryophyte | 511 | 25 | 8 | 9 | 8 |
| Selaginella moellendorffii | Spike moss | Lycophyte | 100 | 2 | 0 | NA | NA |
| Arabidopsis thaliana | Thale cress | Dicot | 125 | 151 | 94 | 55 | 0 |
| Carica papaya | Papaya | Dicot | 372 | 34 | 6 | 4 | 1 |
| Oryza sativa | Rice | Monocot | 466 | 458 | 0 | 274 | 182 |
| Vitis vinifera | Wine grape | Dicot | 487 | 459 | 97 | 215 | 147 |
| Zea mays | Maize | Monocot | 2400 | 95 | 0 | 71 | 23 |
| Triticum aestivum | Bread wheat | Monocot | ~17,000 | >2000 | 0 | NA | NA |
This expansion is not linearly correlated with genome size, as demonstrated by bladderwort (Utricularia gibba), where NLRs constitute only 0.003% of all coding genes, compared to 2% in apple (Malus domestica) [2]. The variation in NLR numbers across species suggests lineage-specific evolutionary trajectories driven by distinct pathogen pressures and ecological niches.
Evolutionary Mechanisms Driving NLR Diversity
NLR gene families evolve through several genetic mechanisms that generate diversity while maintaining functional integrity:
Tandem duplication: This represents a primary driver of NLR family expansion, particularly in flowering plants. In pepper (Capsicum annuum), tandem duplication accounts for 18.4% (53/288) of NLR genes, with notable clustering on chromosomes 08 and 09 [42]. These genomic regions, often near telomeres with higher recombination frequencies, serve as hotspots for rapid generation of novel resistance specificities [30] [42].
Segmental duplication and polyploidization: Whole-genome duplication events contribute significantly to NLR expansion, particularly in species with recent polyploidization history. Hexaploid wheat (Triticum aestivum) contains over 2,000 NLR genes—the largest reported repertoire—though subsequent pseudogenization often follows initial expansion [30].
Diversifying selection: Positive selection acts preferentially on solvent-exposed residues of the leucine-rich repeat (LRR) domains, facilitating adaptation to evolving pathogen effectors [30]. This diversifying selection enables LRR domains to evolve novel binding specificities despite structural conservation.
Domain shuffling and recombination: The modular architecture of NLR proteins facilitates domain rearrangements, allowing the evolution of new recognition specificities. The emergence of integrated domains that mimic pathogen targets (decoys) represents a key innovation in NLR evolution [2].
The distribution of NLR subclasses also reveals evolutionary patterns. Toll/interleukin-1 receptor (TIR)-type NLRs (TNLs) are largely absent from monocots, while coiled-coil (CC)-type NLRs (CNLs) are present in both monocots and dicots [30]. Some dicots exhibit unbalanced ratios; Brassicaceae species have approximately twice as many TNLs as CNLs, while Solanaceae species like potato and grapevine show the opposite pattern with CNLs outnumbering TNLs 4:1 [30].
Molecular Mechanisms of NLR Regulation and Autoimmunity
Structural Basis of NLR Auto inhibition and Activation
Plant NLRs function as molecular switches that transition between inactive and active states through nucleotide-dependent conformational changes. In the absence of pathogens, NLRs maintain an auto inhibited state bound to ADP, with the LRR domain stabilizing this inactive conformation [30] [41]. Recent structural studies have revealed sophisticated mechanisms that maintain this quiescent state:
Oligomerization-mediated autoinhibition: The tomato NLR protein NRC2 (SlNRC2) forms dimers, tetramers, and higher-order oligomers that stabilize its inactive conformation. Cryo-electron microscopy structures demonstrate that these oligomeric interfaces sequester SlNRC2 from assembling into active complexes, with mutations at dimeric or interdimeric interfaces enhancing pathogen-induced cell death and immunity [43].
Cofactor binding: Structural analyses unexpectedly revealed inositol hexakisphosphate (IP6) or pentakisphosphate (IP5) bound to the inner surface of the C-terminal LRR domain of SlNRC2. Mutations at this inositol phosphate-binding site impair SlNRC2-mediated cell death, suggesting these molecules serve as essential cofactors for NLR function [43].
Conformational equilibrium: Rather than a simple binary switch, NLRs exist in an equilibrium between ON and OFF states. Pathogen effectors bind to and stabilize the ON state, shifting this equilibrium toward active conformations that trigger defense signaling [30].
Diagram: NLR Activation and Regulatory Mechanisms
Expression Regulation and Its Consequences
Tight regulation of NLR expression is essential to prevent autoimmunity while maintaining preparedness for pathogen attack. Multiple regulatory layers operate to control NLR abundance:
Transcriptional regulation: NLR promoters are enriched in defense-related cis-regulatory elements, with 82.6% of pepper NLR promoters containing binding sites for salicylic acid (SA) and/or jasmonic acid (JA) signaling [42]. This enables coordinated induction during immune responses while maintaining baseline expression.
MicroRNA-mediated control: Numerous microRNAs target conserved NLR motifs (e.g., P-loop) in flowering plants, providing bulk control of NLR transcripts that may allow plant species to maintain large NLR repertoires without deleterious effects [1].
Expression level signatures: Contrary to the historical assumption that NLRs are transcriptionally repressed, functional NLRs actually exhibit high steady-state expression levels in uninfected plants. Known functional NLRs are significantly enriched in the top 15% of expressed NLR transcripts across multiple species, suggesting that adequate expression is prerequisite for function rather than necessarily leading to autoimmunity [35].
The fitness costs of improper NLR regulation are substantial. In Arabidopsis thaliana, certain allelic combinations of NLR genes DM1 and DM2 cause autoimmunity in hybrid offspring, demonstrating how regulatory incompatibilities can arise from natural variation [30]. Similarly, the presence of the Arabidopsis NLR RPM1 reduces silique and seed production, while overexpression of RPW8 and LAZ5 causes spontaneous cell death even in the absence of pathogens [35].
Experimental Approaches for Studying NLR-Mediated Immunity and Fitness Costs
Methodologies for NLR Functional Characterization
Comprehensive understanding of NLR biology requires integrated experimental approaches that span molecular, genomic, and phenotypic analyses:
Table 2: Key Experimental Protocols for NLR Research
| Method Category | Specific Protocol | Key Applications | Technical Considerations | |
|---|---|---|---|---|
| Genome-wide Identification | HMMER domain search (PF00931) | Identification of NLR repertoires in sequenced genomes | E-value cutoff 1×10⁻⁵; validation via NCBI CDD (cd00204) and Pfam [42] | |
| Expression Analysis | RNA-Seq differential expression | Identification of NLRs responsive to pathogen infection | FDR < 0.05, |log₂FC| ≥ 1; tissue-specific considerations crucial [42] [35] | |
| Functional Validation | High-throughput transformation | Large-scale NLR functional screening | Wheat transgenic array of 995 NLRs; requires efficient transformation system [35] | |
| Protein Interaction Studies | Yeast two-hybrid/Co-IP | Mapping NLR interaction networks | Identification of helper/sensor partnerships; confirmation of resistosome formation [43] | |
| Structural Biology | Cryo-electron microscopy | Determining NLR atomic structures | Reveals autoinhibition mechanisms, cofactor binding, oligomeric states [43] |
Research Reagent Solutions for NLR Studies
Table 3: Essential Research Tools for NLR Investigation
| Reagent/Resource | Function | Application Examples |
|---|---|---|
| NB-ARC domain HMM profile (PF00931) | Bioinformatics identification | Core domain recognition in genome annotations [42] |
| PlantCARE database | cis-regulatory element prediction | Identification of defense-related promoter motifs [42] |
| STRING database | Protein-protein interaction prediction | Mapping NLR immune networks [42] |
| SWISS-MODEL | Protein structure prediction | Homology modeling of NLR domains [42] |
| Dual Synteny Plotter (TBtools) | Comparative genomics | Identification of orthologous NLR clusters [42] |
| High-efficiency wheat transformation | Functional validation | Large-scale transgenic array generation [35] |
Recent innovative approaches have enabled significant advances in NLR functional characterization. A notable example is the development of a pipeline that combines expression signature analysis with high-throughput transformation, enabling screening of 995 NLRs from diverse grass species for resistance to wheat stem rust and leaf rust pathogens. This approach identified 31 new resistance NLRs (19 against stem rust, 12 against leaf rust), demonstrating the power of large-scale functional screening [35].
Diagram: Experimental Workflow for NLR Functional Analysis
Balancing Immunity and Fitness: Evolutionary Perspectives and Future Directions
The conundrum of maintaining effective immunity without debilitating autoimmunity has shaped NLR evolution across land plants. Several key principles emerge from comparative analyses:
First, the metabolic costs of NLR-mediated immunity manifest through multiple mechanisms. These include the direct costs of synthesizing and maintaining NLR proteins, the energetic expenditure of immune activation, and the potential yield penalties from inadvertent autoimmunity [30] [41]. These costs create selective pressures that fine-tune NLR regulation across evolutionary timescales.
Second, the solution to this balancing act varies across plant lineages. Long-lived woody species like apple have expanded NLR repertoires (nearly 1,000 genes) that may compensate for infrequent meiosis and limited capacity to generate novelty through sexual recombination [30]. In contrast, annual species with shorter generation times may rely more heavily on rapid sequence diversification and selective sweeps.
Third, the helper NLR system in Solanaceae represents an evolutionary innovation that optimizes the balance between recognition diversity and signaling efficiency. Rather than maintaining numerous complete signaling pathways, plants deploy a limited set of highly expressed helper NLRs (NRCs) that interface with diverse sensor NLRs [43]. This architecture reduces the fitness costs associated with maintaining multiple redundant signaling pathways while expanding recognition capacity.
Future research directions should focus on elucidating how NLR expression thresholds influence immune activation and autoimmunity, particularly in the context of changing environmental conditions. The discovery that functional NLRs tend to be highly expressed challenges longstanding paradigms and suggests new strategies for engineering disease resistance in crops [35]. Furthermore, understanding how NLR networks rather than individual genes contribute to the balance between immunity and fitness will be essential for predicting the durability of resistance genes in agricultural systems.
As climate change alters pathogen distributions and plant-pathogen interactions, understanding the evolutionary principles that balance immunity and fitness becomes increasingly crucial. The insights gained from studying NLR diversity and regulation across plant evolution provide a foundation for developing sustainable crop protection strategies that maximize resistance while minimizing fitness costs—a critical imperative for global food security.
In plants, nucleotide-binding domain and leucine-rich repeat (NLR) proteins function as sophisticated intracellular immune receptors that mediate effector-triggered immunity (ETI), often culminating in a hypersensitive response (HR) characterized by programmed cell death at the infection site [30]. The evolution of NLR genes spans over one billion years, originating in green plants and diverging into at least three major subclasses: TIR-NLRs (TNLs), CC-NLRs (CNLs), and RPW8-NLRs (RNLs) [3]. This gene family has undergone massive expansion in flowering plants, with NLR repertoires ranging from approximately 80 members in Brassica rapa to over 450 in wine grape (Vitis vinifera), in stark contrast to the modest 25 NLRs found in the bryophyte Physcomitrella patens and a mere 2 in the lycophyte Selaginella moellendorffii [1] [44].
This dramatic lineage-specific expansion reflects a continuous evolutionary arms race with rapidly evolving pathogens, but it comes with significant fitness costs. Uncontrolled NLR activation leads to autoimmune phenotypes, retarded growth, and yield penalties [30] [45]. Consequently, plants have evolved multi-layered regulatory mechanisms to maintain NLRs in a tightly controlled yet rapidly inducible state. These mechanisms operate at transcriptional, post-transcriptional, and allosteric levels and have co-evolved with the NLR family itself across land plants, from early diverging lineages like mosses to highly specialized dicots. Understanding these control mechanisms provides crucial insights into the evolutionary constraints shaping plant immune systems and offers strategies for engineering disease-resistant crops without compromising fitness.
Transcriptional Regulation of NLR Genes
Epigenetic Control Through Poised Chromatin States
Recent research has revealed that epigenetic mechanisms, particularly specialized chromatin states, maintain NLR genes in a transcriptionally primed but suppressed condition under non-stress conditions. In soybean (Glycine max), integrative epigenomic and transcriptomic analyses have identified that both NLR and pattern recognition receptor (PRR) genes harbor bivalent chromatin modifications, characterized by the simultaneous presence of active histone marks (H3K4me3, H3K27ac, H3K9ac, H4K16ac) and repressive marks (H3K27me3) [46]. This poised state maintains low basal expression while enabling rapid transcriptional activation upon pathogen perception.
Distinct epigenetic features differentiate NLR regulatory patterns: NLR genes display narrow H3K27me3 peaks with strong RNA Polymerase II pausing at their 5' ends, whereas PRR genes exhibit broader H3K27me3 domains [46]. This pronounced Pol II pausing at NLR promoters facilitates rapid transcriptional elongation upon receiving activation signals. Furthermore, clustered NLR and PRR genes residing within the same topologically associating domains share similar chromatin states and expression dynamics, suggesting coordinated epigenetic control of immune gene clusters [46].
Table 1: Histone Modifications Associated with Poised Chromatin States in Plant Immune Genes
| Histone Modification | Type | Function in Transcriptional Poising |
|---|---|---|
| H3K4me3 | Active | Marks promoters of transcriptionally ready genes |
| H3K27ac | Active | Associated with active enhancers and promoters |
| H3K9ac | Active | Correlates with transcriptional competence |
| H4K16ac | Active | Promotes chromatin accessibility |
| H3K27me3 | Repressive | Maintains transcriptional repression at poised genes |
| H3K36me2/3 | Active | Associated with transcriptional elongation |
Cis-Regulatory Elements and Transcription Factors
NLR gene promoters are enriched with defense-related cis-regulatory elements that respond to hormonal signaling pathways, particularly salicylic acid (SA) and jasmonic acid (JA). In pepper (Capsicum annuum), 82.6% of NLR promoters (238 out of 288 genes) contain binding sites for SA and/or JA signaling components [42]. These regulatory motifs serve as integration points for defense signaling pathways, allowing coordinated expression of NLR networks in response to pathogen attack.
The chromosomal distribution of NLR genes further influences their transcriptional regulation. NLRs frequently cluster in subtelomeric regions with high recombination frequencies, as observed in common bean (Phaseolus vulgaris), potato (Solanum tuberosum), tomato (Solanum lycopersicum), and foxtail millet (Setaria italica) [30]. These genomic locations facilitate rapid evolution of new recognition specificities while potentially enabling coordinated transcriptional control of clustered NLR families through shared regulatory elements.
Post-transcriptional Regulation Mechanisms
Alternative Splicing and Polyadenylation
Alternative splicing generates multiple transcript variants from a single NLR gene, increasing proteomic diversity and providing regulatory potential. This process can produce truncated protein isoforms that function as dominant-negative regulators or fine-tune immune signaling outputs [45]. Similarly, alternative polyadenylation site selection generates NLR transcripts with varying 3'UTR lengths, influencing their stability, localization, and translation efficiency under different physiological conditions.
Small RNA-Mediated Regulation
MicroRNAs (miRNAs) play a crucial role in post-transcriptional control of NLR genes. Numerous miRNAs target conserved nucleotide sequences encoding NLR motifs, such as the P-loop, in flowering plants [1] [19]. This bulk control of NLR transcripts may allow plant species to maintain extensive NLR repertoires without depletion of functional NLR loci or incurring excessive fitness costs [1]. The co-evolution of NLR gene families and miRNA regulatory networks represents an important mechanism for balancing immune responsiveness with growth requirements.
Nonsense-Mediated Decay Surveillance
Nonsense-mediated decay (NMD) pathways surveil NLR transcripts and degrade those containing premature termination codons, which frequently arise from alternative splicing or genomic mutations [45]. This quality control mechanism prevents the accumulation of potentially deleterious truncated NLR proteins that could cause constitutive immune activation or interfere with proper immune signaling.
Table 2: Post-transcriptional Regulatory Mechanisms for NLR Genes
| Mechanism | Molecular Basis | Functional Outcome |
|---|---|---|
| Alternative Splicing | Generation of multiple mRNA isoforms from a single gene | Produces regulatory variants; increases immune receptor diversity |
| miRNA-mediated repression | miRNA binding to complementary NLR mRNA sequences | Fine-tunes NLR expression levels; reduces fitness costs |
| Nonsense-Mediated Decay (NMD) | Degradation of transcripts with premature termination codons | Prevents accumulation of aberrant NLR proteins |
| Alternative Polyadenylation | Selection of different 3' end processing sites | Modifies transcript stability and translation efficiency |
Allosteric Regulation and Protein-Level Control
Nucleotide-Dependent Molecular Switching
NLR proteins function as nucleotide-operated molecular switches that cycle between ADP-bound (off) and ATP-bound (on) states. In the absence of pathogens, NLRs maintain an auto-inhibited conformation with ADP bound to the NB-ARC domain, stabilized by intramolecular interactions with the LRR domain [30]. The current model proposes that NLRs exist in an equilibrium between inactive and active states, with effector binding shifting this equilibrium toward the active conformation [30].
Upon effector recognition, conformational changes in the LRR domain trigger nucleotide exchange (ADP to ATP) in the NB-ARC domain, leading to NLR activation and immune signaling initiation [30]. This conserved switch mechanism represents a fundamental allosteric control system that has been maintained throughout NLR evolution across land plants.
Oligomerization and Resistosome Formation
Activated NLR proteins undergo oligomerization to form higher-order complexes known as resistosomes. Structural studies have revealed that CNLs like ZAR1 from Arabidopsis thaliana and Sr35 from wheat form calcium-permeable cation channels upon oligomerization [3]. These resistosomes directly couple immune recognition to downstream signaling by mediating calcium influx into the cytoplasm, which serves as a secondary messenger for defense activation.
The oligomerization process is itself subject to allosteric control, as demonstrated by the rice Pik NLR pair. Single amino acid polymorphisms at the interaction interface between sensor (Pik-1) and helper (Pik-2) NLRs determine their preferential association, ensuring proper immune activation while preventing dangerous autoimmunity in mismatched pairs [47]. This specificity highlights how allosteric controls have co-evolved in genetically linked NLR pairs to maintain immune homeostasis.
Helper-Sensor NLR Networks
Many NLRs function within cooperative networks where sensor NLRs (typically TNLs or CNLs) detect pathogen effectors and require helper NLRs (often RNLs) for signal transduction. In seed plants, TNL activation requires the EDS1 (Enhanced Disease Susceptibility 1) family proteins, which form heterodimers with helper RNLs to transduce immune signals [3]. This division of labor creates built-in allosteric control points, as proper immune activation requires coordinated conformational changes in both sensor and helper components.
The co-evolution of sensor and helper NLR families has shaped their functional specialization. For instance, the loss of TNLs in grasses correlates with the absence of EDS1 family members, demonstrating how signaling components and NLR subtypes have co-evolved [21]. This coordinated evolutionary history ensures that allosteric control mechanisms remain effective despite rapid sequence diversification in pathogen recognition domains.
Experimental Protocols for Studying NLR Regulation
Epigenomic Profiling of NLR Chromatin States
Purpose: To characterize poised chromatin states at NLR loci and their dynamics during immune activation.
Methodology:
- Perform Chromatin Immunoprecipitation sequencing (ChIP-seq) for histone modifications (H3K4me3, H3K27me3, H3K27ac, H3K9ac, H4K16ac, H3K36me2/3) using cross-linked chromatin from untreated and immunologically stimulated tissues.
- Conduct Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) to map open chromatin regions.
- Perform RNA Polymerase II ChIP-seq with antibodies specific for Ser2P (elongating Pol II) and Ser5P (initiating Pol II).
- Integrate datasets using computational approaches like ChromHMM to define chromatin states genome-wide.
- Validate findings through targeted mutagenesis of cis-regulatory elements and monitoring of NLR expression changes.
Key Considerations: Include biological replicates with high sequencing depth (>50 million reads per sample) for robust peak calling. Normalize histone modification signals to input controls and compare to known benchmark regions [46].
Assessing NLR Activation Through Cell Death Assays
Purpose: To quantify NLR-mediated immune responses and autoimmunity in matched versus mismatched NLR pairs.
Methodology:
- Clone NLR genes into binary expression vectors under strong promoters (e.g., 35S).
- Express matched and mismatched NLR combinations in Nicotiana benthamiana leaves via Agrobacterium-mediated transient transformation.
- Include positive and negative controls: known autoimmune combinations and empty vector controls.
- Monitor hypersensitive response symptoms over 2-5 days post-infiltration.
- Quantify cell death using electrolyte leakage measurements or Evans Blue staining.
- Confirm protein expression and interaction through immunoblotting and co-immunoprecipitation.
Key Considerations: Use multiple allelic variants to assess specificity. Titrate Agrobacterium densities to ensure comparable expression levels across samples [47].
Identification and Evolutionary Analysis of NLR Genes
Purpose: To conduct genome-wide identification, classification, and evolutionary analysis of NLR gene families.
Methodology:
- Perform HMMER searches against target proteomes using NB-ARC domain hidden Markov models (PF00931) with E-value cutoff of 1×10⁻⁵.
- Validate candidates through NCBI Conserved Domain Database (cd00204) and Pfam batch searches.
- Classify NLRs into TNL, CNL, and RNL subclasses based on N-terminal domains (TIR, CC, RPW8).
- Identify orthogroups across species using OrthoFinder with Diamond for sequence alignment and MCL for clustering.
- Analyze duplication events through MCScanX synteny analysis.
- Construct phylogenetic trees using Maximum Likelihood methods (IQ-TREE) with 1000 bootstrap replicates.
Key Considerations: Manually inspect domain architectures to distinguish functional NLRs from truncated forms or pseudogenes. Use established reference NLR sets from well-annotated genomes (e.g., Arabidopsis thaliana) for comparison [19] [42].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for NLR Regulation Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Antibodies for Epigenomics | Anti-H3K4me3, Anti-H3K27me3, Anti-H3K27ac, Anti-Ser2P RNA Pol II | Chromatin immunoprecipitation to map histone modifications and transcriptional activity |
| Cloning Systems | Gateway-compatible vectors, Golden Gate modular systems, 35S promoters | Efficient construction of NLR expression clones for functional assays |
| Transformation Systems | Agrobacterium tumefaciens GV3101, LBA4404 | Transient or stable plant transformation for functional studies |
| Cell Death Markers | Evans Blue, electrolyte leakage measurement kits | Quantification of hypersensitive cell death responses |
| Domain Databases | Pfam (NB-ARC: PF00931), NCBI CDD (cd00204) | Identification and annotation of NLR domains in genomic sequences |
| Bioinformatics Tools | OrthoFinder, MCScanX, ChromHMM, PlantCARE | Evolutionary analysis, synteny mapping, chromatin state definition, promoter element identification |
Visualization of NLR Regulatory Networks
Concluding Perspectives
The multi-layered regulatory mechanisms controlling NLR genes represent evolutionary solutions to the fundamental challenge of maintaining effective immunity while minimizing fitness costs. The emergence of poised chromatin states in vascular plants, the co-evolution of miRNA regulatory networks in flowering plants, and the functional specialization of NLR pairs across plant lineages all reflect adaptive innovations that shape today's plant immune systems.
Future research directions should include comparative epigenomic studies across diverse plant species to understand how chromatin-based regulation has evolved in different phylogenetic contexts, structural studies of NLR allosteric transitions to inform engineering approaches, and investigation of how regulatory mechanisms integrate with recently discovered NLR signaling components. Such efforts will continue to reveal the sophisticated balancing act plants perform to survive in a pathogen-rich world, providing insights that could transform crop improvement strategies.
Nucleotide-binding leucine-rich repeat (NLR) genes constitute the largest and most critical family of plant disease resistance genes, encoding intracellular immune receptors that recognize pathogen effectors and initiate robust defense responses. The genomic copy number of these genes varies tremendously across plant species, creating a complex landscape of dosage requirements for effective immunity. This technical guide explores the evolutionary patterns, structural classifications, and functional constraints that govern NLR copy number thresholds in land plants. By integrating findings from recent large-scale genomic studies across species ranging from mosses to dicots, we examine how ecological specialization, whole-genome duplication events, and tandem gene duplications have shaped NLR repertoires. The synthesis of phylogenomic, synteny, and expression analyses presented herein provides a framework for understanding the dosage sensitivity of NLR genes and their application in developing disease-resistant crops.
Plant immunity relies on a sophisticated surveillance system where NLR (Nucleotide-binding Leucine-Rich Repeat) proteins function as intracellular immune receptors that detect pathogen-derived molecules and initiate effector-triggered immunity (ETI) [48]. These proteins typically exhibit a modular architecture consisting of three fundamental domains: an N-terminal signaling domain (either TIR, CC, or RPW8), a central nucleotide-binding adaptor shared with APAF-1, R proteins, and CED-4 (NB-ARC) domain, and a C-terminal leucine-rich repeat (LRR) region responsible for pathogen recognition [19] [49]. Based on their N-terminal domains, NLRs are classified into three major subclasses: TIR-NLRs (TNLs), CC-NLRs (CNLs), and RPW8-NLRs (RNLs), each with distinct signaling mechanisms and evolutionary trajectories [49] [21].
The copy number of NLR genes in plant genomes exhibits remarkable variation, ranging from just a few in green algae to over 2,000 in hexaploid wheat (Triticum aestivum) [19] [49]. This variation reflects a dynamic evolutionary arms race between plants and their pathogens, driven by continuous selective pressure to recognize rapidly evolving pathogen effectors. Recent studies have revealed that NLR copy number thresholds are not arbitrary but represent optimized genomic configurations shaped by ecological adaptation, life history strategies, and molecular constraints on protein function [21] [7]. Understanding these dosage requirements is essential for harnessing NLR genes in crop improvement and sustainable agriculture.
Evolutionary Dynamics of NLR Genes Across Land Plants
Phylogenetic Distribution and Diversification
Comprehensive genomic analyses across diverse plant lineages have revealed dramatic fluctuations in NLR gene copy numbers throughout plant evolution. While bryophytes like Physcomitrella patens possess relatively small NLR repertoires of approximately 25 genes, flowering plants have experienced substantial gene family expansion, with some species containing hundreds to thousands of NLR copies [19]. A recent angiosperm NLR atlas (ANNA) analyzing over 300 angiosperm genomes demonstrated that NLR copy numbers can differ up to 66-fold among closely related species due to rapid gene loss and gain events [21].
Table 1: NLR Gene Distribution Across Representative Plant Species
| Plant Species | Lineage | Total NLRs | CNLs | TNLs | RNLs | Specialization |
|---|---|---|---|---|---|---|
| Physcomitrella patens | Moss | ~25 | Not specified | Not specified | Not specified | Baseline NLR repertoire |
| Oropetium thomaeum | Monocot (Poaceae) | Few dozen | Majority | 0 | Few | Extreme NLR contraction |
| Triticum aestivum | Monocot (Poaceae) | >2,000 | Majority | 0 | Few | Significant NLR expansion |
| Angelica sinensis | Dicot (Apiaceae) | 95 | 79 | 14 | 2 | Medicinal plant, NLR reduction |
| Coriandrum sativum | Dicot (Apiaceae) | 183 | 148 | 31 | 4 | Culinary herb, moderate NLR expansion |
| Arabidopsis thaliana | Dicot (Brassicaceae) | ~200 | ~150 | ~50 | ~4 | Model plant, balanced repertoire |
| Gossypium hirsutum (Mac7) | Dicot (Malvaceae) | Not specified | Not specified | Not specified | Not specified | CLCuD tolerant, high NLR diversity |
Several key evolutionary patterns have emerged from comparative genomic studies:
Convergent NLR reduction is associated with adaptations to specialized ecological niches, particularly aquatic, parasitic, and carnivorous lifestyles [21]. The independent NLR contraction observed in multiple aquatic plant lineages mirrors the limited NLR expansion in green algae prior to terrestrial colonization.
Differential retention of NLR subclasses has occurred throughout plant evolution, most notably exemplified by the complete loss of TNL genes in monocots, despite their persistence in eudicots [49]. Compelling microsynteny evidence indicates a clear correspondence between non-TNLs in monocots and the extinct TNL subclass, suggesting functional compensation rather than simple gene loss [49].
Co-evolution between NLR subclasses and signaling components has been identified, with deficiencies in downstream immune pathway elements potentially driving TNL loss in certain lineages [21]. For instance, the loss of TNLs in monocots may coincide with the absence of their required signal transduction partners [49].
Impact of Whole Genome Duplication and Tandem Duplication
Plant NLR repertoires have been shaped by both small-scale and whole-genome duplication (WGD) events, with different evolutionary consequences for gene dosage. WGD events, such as those observed in the Apioideae subfamily of Apiaceae, initially produce duplicated NLR copies that may be retained due to reduced selective pressure [7]. However, these duplicates are frequently followed by extensive gene loss and subfunctionalization over evolutionary time [19] [7].
In contrast, tandem duplications represent a primary mechanism for rapid NLR expansion and diversification, often resulting in genomic clusters of NLR genes with variant specificities [19]. A study of 12,820 NBS-domain-containing genes across 34 plant species identified numerous tandem duplication events, particularly within specific orthogroups associated with disease resistance [19]. These tandem arrays create hotspots for NLR sequence evolution through gene conversion and unequal crossing over, generating novel pathogen recognition capabilities while maintaining core NLR functions.
Figure 1: Evolutionary Pathways Shaping NLR Copy Number in Plants. NLR gene families experience dynamic expansion through whole genome and tandem duplication events, followed by contraction through gene loss, with outcomes influenced by ecological adaptation and functional selection.
Classification Systems and Structural Diversity of NLR Genes
Advanced Classification Based on Synteny and Phylogenetics
Traditional NLR classification based solely on N-terminal domains has been refined through integrated synteny and phylogenetic analyses. A novel classification system for angiosperm NLR genes, grounded in network analysis of microsynteny information, categorizes these genes into five distinct classes: CNLA, CNLB, CNL_C, TNL, and RNL [49]. This refined classification reveals previously unrecognized evolutionary relationships and functional specializations within the NLR superfamily.
The credibility of this five-class system is supported by both phylogenetic analysis and examination of protein domain structures [49]. Well-characterized CNL genes display distinct distributions across the three CNL subclasses: GmRps1k, OsXa1, OsR3, and SlI2 cluster within CNLA; AtZAR1, AtLOV1, TaSr35, OsPi9, SlPRF, SlSw5b, and NbNRC2b/4b group in CNLB; and AtSUMM2, AtRPS5, and AtRPS2 belong to CNL_C [49]. This refined classification provides a more accurate framework for understanding NLR gene evolution, expression patterns, and functional capabilities.
Table 2: NLR Subclassification Based on Synteny and Phylogenetic Analysis
| NLR Class | Representative Members | Structural Features | Evolutionary Patterns |
|---|---|---|---|
| CNL_A | GmRps1k, OsXa1, OsR3, SlI2 | CC domain, NBS, LRR | Diversified pathogen recognition |
| CNL_B | AtZAR1, AtLOV1, TaSr35, OsPi9, SlPRF, SlSw5b, NbNRC2b/4b | CC domain, NBS, LRR | Forms cation-channel resistosomes |
| CNL_C | AtSUMM2, AtRPS5, AtRPS2 | CC domain, NBS, LRR | Sister group to CNLA and CNLB |
| TNL | Multiple members across dicots | TIR domain, NBS, LRR | Lost in monocots; forms NADase resistosomes |
| RNL | ADR1, NRG1 | RPW8 domain, NBS, LRR | Helper NLRs; minimal expansion |
Domain Architecture Diversity
NLR genes exhibit remarkable diversity in their domain architectures, extending beyond the canonical NBS-LRR structure. A comprehensive analysis of 12,820 NBS-domain-containing genes across 34 plant species identified 168 distinct classes with several novel domain architecture patterns [19]. These include both classical structures (NBS, NBS-LRR, TIR-NBS, TIR-NBS-LRR) and species-specific structural patterns (TIR-NBS-TIR-Cupin1-Cupin1, TIR-NBS-Prenyltransf, Sugar_tr-NBS) [19].
This architectural diversity likely reflects functional specialization and adaptation to recognize specific pathogen classes. Integrated domain fusions, where additional protein domains are incorporated into NLR proteins, can directly participate in pathogen recognition or modulate signaling output [19]. The functional validation of these diverse architectures remains an active area of research, with implications for engineering novel disease resistance specificities in crop plants.
Methodological Framework for NLR Gene Analysis
Genomic Identification and Annotation Pipeline
Accurate identification and annotation of NLR genes is fundamental to copy number analysis. The following integrated protocol combines multiple computational approaches for comprehensive NLR characterization:
Software Installation and Requirements
- Operating System: 64-bit Linux or Mac OS X
- NLRtracker v1.0.3: For sensitive NLR annotation from protein sequences [48]
- NLR-Annotator v2.1: Alternative for nucleotide sequence input [48]
- InterProScan 5.53-87.0: Protein function characterization [48]
- MAFFT v7: Multiple sequence alignment [48]
- RAxML v8.2.12: Maximum likelihood phylogenetic inference [48]
- MEME Suite v5.5.5: Motif-based sequence analysis [48]
- BLAST+ v2.12.0: Local sequence similarity searches [48]
- MCL v14-137: Network clustering for orthogroup identification [48]
Step-by-Step Protocol
- Data Acquisition: Download protein sequence files from reference genome databases (e.g., Phytozome, NCBI, Plaza) [19] [48].
- NLR Annotation: Annotate NLRs from input protein sequences using NLRtracker:
./NLRtracker -s input_protein.fasta -o NLRtracker_output[48]. - Domain Verification: Verify NBS (NB-ARC) domains using HMMER search against Pfam database (PF00931) with E-value cutoff of 10⁻⁴ [7].
- Orthogroup Delineation: Cluster NLR genes into orthogroups using OrthoFinder v2.5.1 with DIAMOND for sequence similarity searches and MCL for clustering [19].
- Motif Analysis: Identify conserved sequence motifs using MEME Suite with standard parameters [48].
Figure 2: Computational Workflow for NLR Gene Identification and Evolution Analysis. The pipeline integrates multiple bioinformatic tools for comprehensive characterization of NLR gene families from genomic data.
Expression Analysis and Functional Validation
Transcriptomic profiling of NLR genes across tissues and stress conditions provides critical insights into their functional roles and dosage sensitivity. Standardized methodology includes:
- Data Collection: Retrieve RNA-seq data from specialized databases (IPF database, Cotton Functional Genomics Database, CottonGen database, NCBI BioProjects) [19].
- Expression Quantification: Calculate FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values and categorize into tissue-specific, abiotic stress-specific, and biotic stress-specific expression profiles [19].
- Differential Expression: Identify significantly upregulated NLR orthogroups under pathogen challenge using appropriate statistical thresholds.
- Functional Validation: Implement virus-induced gene silencing (VIGS) to test putative NLR functions, as demonstrated for GaNBS (OG2) in resistant cotton, which confirmed its role in virus titrating [19].
For genetic variation analysis, compare SNP and indel profiles between susceptible and tolerant accessions, such as the 6583 unique variants identified in tolerant Mac7 versus 5173 variants in susceptible Coker312 Gossypium hirsutum accessions [19]. Protein-ligand and protein-protein interaction studies can further validate NLR functions through demonstrated interactions with ADP/ATP and core viral proteins [19].
Table 3: Key Research Reagents and Computational Tools for NLR Studies
| Resource Category | Specific Tool/Reagent | Application | Key Features |
|---|---|---|---|
| Annotation Tools | NLRtracker v1.0.3 | NLR identification from protein sequences | High sensitivity for diverse plant species |
| NLR-Annotator v2.1 | NLR annotation from nucleotide sequences | Handles genomic DNA sequences | |
| Domain Analysis | InterProScan 5.53-87.0 | Protein domain characterization | Integrates multiple domain databases |
| HMMER v3.4 | Sequence homology searches | Profile hidden Markov models | |
| Phylogenetic Analysis | MAFFT v7 | Multiple sequence alignment | Handles large datasets efficiently |
| RAxML v8.2.12 | Phylogenetic tree construction | Maximum likelihood method | |
| IQ-TREE | Phylogenetic inference | Model selection integration | |
| Motif Discovery | MEME Suite v5.5.5 | Conserved motif identification | Web interface or command line |
| Orthology Analysis | OrthoFinder v2.5.1 | Orthogroup delineation | Gene duplication event inference |
| MCL v14-137 | Network clustering | Orthogroup refinement | |
| Expression Analysis | Various RNA-seq databases | Expression profiling | Tissue/stress-specific data |
| Functional Validation | VIGS constructs | Gene silencing | Functional confirmation in planta |
| Yeast two-hybrid systems | Protein interaction studies | NLR-pathogen effector interactions |
The copy number thresholds governing NLR function represent a complex interplay between evolutionary history, ecological adaptation, and molecular constraints. The dynamic nature of NLR gene families—characterized by rapid expansion and contraction across plant lineages—underscores their crucial role in plant-pathogen coevolution. The refined classification systems emerging from synteny-informed phylogenomics provide unprecedented resolution for understanding NLR gene evolution and functional specialization.
Future research directions should focus on several key areas:
- Mechanistic Basis of Dosage Sensitivity: Elucidating the molecular mechanisms that make certain NLR genes dosage-sensitive while others tolerate copy number variation.
- Engineering Optimal NLR Dosage: Leveraging synthetic biology approaches to design NLR gene clusters with enhanced disease resistance spectra without fitness costs.
- Cross-Species Comparative Analyses: Expanding NLR atlas initiatives to encompass broader phylogenetic diversity, including non-model species with unique ecological adaptations.
- Single-Cell Expression Atlas: Developing tissue-specific and cell-type-specific NLR expression profiles to understand spatial aspects of NLR dosage requirements.
The continued integration of evolutionary genomics, molecular biology, and computational approaches will further refine our understanding of NLR copy number thresholds and accelerate the development of durable disease resistance in crop plants.
Overcoming Silencing and Instability in Transgenic NLR Stacking
The evolution of Nucleotide-binding Leucine-rich Repeat (NLR) genes in land plants represents a dynamic arms race between hosts and their pathogens. From the limited NLR repertoires in mosses (e.g., ~25 in Physcomitrella patens) to the massive expansions in dicots (e.g., up to 459 in wine grape), the story of NLRs is one of continual innovation and diversification [1] [2]. This evolutionary history, characterized by frequent gene duplication, neofunctionalization, and birth-and-death dynamics, provides the fundamental justification for deploying NLR stacks in transgenic approaches for durable disease resistance [2] [50]. NLRs are the cornerstone of Effector-Triggered Immunity (ETI), providing a robust, often hypersensitive response (HR)-associated defense that is highly specific [1] [37]. However, the deployment of single NLR transgenes in agriculture is frequently overcome by rapidly evolving pathogen populations. Stacking multiple NLRs into a single plant line presents a formidable barrier to pathogens, mimicking the complex NLR networks that have evolved naturally in many plant species [51] [37]. Despite the promise of this strategy, the transgenic stacking of NLRs is fraught with technical challenges, primarily stemming from unintended silencing and structural instability, which this guide aims to address.
Evolutionary Trajectory of NLR Genes in Land Plants
The NLR immune receptor family has undergone significant expansion and diversification since plants colonized land. The genomic data from extant species reveals a clear evolutionary trajectory.
Table 1: NLR Repertoire Expansion in Land Plants [1]
| Species | Common Name | Clade | Genome Size (Mbp) | Total NLRs | TNLs | CNLs |
|---|---|---|---|---|---|---|
| Physcomitrella patens | Moss | Bryophyte | 511 | 25 | 8 | 9 |
| Selaginella moellendorffii | Spike Moss | Lycophyte | 100 | 2 | 0 | NA |
| Arabidopsis thaliana | Thale Cress | Eudicot | 125 | 151 | 94 | 55 |
| Glycine max | Soybean | Eudicot | 1115 | 319 | 116 | 20 |
| Vitis vinifera | Wine Grape | Eudicot | 487 | 459 | 97 | 215 |
This expansion is not merely quantitative but also qualitative. Early land plants possess NLRs with a wider variety of N-terminal domains, including kinase and α/β hydrolase domains, while angiosperms have largely standardized around Coiled-coil (CC) and Toll/Interleukin-1 Receptor (TIR) N-terminal domains [37]. A key evolutionary development is the emergence of functionally specialized NLR pairs and networks, where sensor NLRs detect pathogen effectors and helper NLRs amplify the immune signal [37]. This functional specialization underscores the logic behind transgenic NLR stacking, aiming to reconstitute complex, resilient immune networks. The high intraspecific diversity and presence/absence variation of NLRs in modern crops are a testament to the intense and ongoing selective pressure exerted by pathogens [2].
Molecular Mechanisms of Silencing and Instability in NLR Stacks
The successful implementation of NLR stacks is hampered by several molecular phenomena that lead to the loss of transgene expression or function.
Transcriptional and Post-transcriptional Silencing
Plants have evolved robust mechanisms to tightly regulate NLR expression, as misexpression can lead to autoimmunity, imposing a fitness cost characterized by reduced growth and yield [52]. These same mechanisms can be aberrantly activated against transgenic NLR stacks.
Transcriptional Gene Silencing (TGS): This involves chromatin-level repression, often triggered by the presence of repetitive DNA sequences, which are common in multi-gene stacks. Key mechanisms include:
- DNA Methylation: Promoter regions of NLRs can be targeted by DNA methylation, leading to transcriptional repression. This is often associated with repeats and transposable elements (TEs) located in promoter regions or introns [52]. For example, the RMG1 TNL in Arabidopsis is methylated on helitron repeats in its promoter, and its induction requires active demethylation [52].
- Histone Modifications: Repressive histone marks, such as H3K9me2, can spread from silenced repetitive elements to adjacent NLR genes. In Arabidopsis, maintaining H3K9me2 on a Copia-type retrotransposon within the first intron of the RPP7 CNL is crucial for its proper transcription and splicing [52].
Post-transcriptional Gene Silencing (PTGS): This involves the degradation of mRNA or inhibition of translation and is a major hurdle for NLR stacks.
- RNA Interference (RNAi): Numerous microRNAs (miRNAs) have been identified that target conserved nucleotide sequences within NLR transcripts, such as the P-loop motif [1]. This is thought to be a natural mechanism to control the cost of NLR expression. In a transgenic context, high expression of repetitive or highly homologous NLR sequences can trigger the production of small interfering RNAs (siRNAs) that direct the sequence-specific degradation of the entire stack's transcripts [52].
Genetic and Structural Instability
The physical arrangement of multiple, often homologous, NLR sequences in a single locus can lead to genomic instability. Homologous recombination between nearly identical sequences can result in intramolecular rearrangements, including deletions, inversions, and gene conversions, which scramble the stack and lead to loss of individual NLRs [2]. This rapid "birth and death" dynamic is a natural feature of NLR cluster evolution but is highly problematic for maintaining designed stacks in transgenic lines.
Strategies to Overcome Silencing and Instability
To counteract these challenges, several advanced molecular strategies can be employed.
Sequence Diversification
A primary strategy is to reduce sequence homology between the stacked NLRs below the threshold that triggers silencing.
- Codon Optimization: Systematically altering the codon usage for each NLR in the stack without changing the amino acid sequence can significantly reduce nucleotide-level homology.
- Synthetic Gene Design: Using protein sequences as a blueprint, design synthetic NLR genes with divergent non-coding regions (introns, UTRs) and exploit the degeneracy of the genetic code to minimize shared siRNA targets, especially in conserved motifs like the P-loop [1].
Chromatin and Epigenetic Engineering
Modifying the transgene locus to create an open chromatin state resistant to silencing is another key approach.
- Matrix Attachment Regions (MARs): Flanking the transgene stack with MARs can insulate it from the repressive effects of the surrounding chromatin and promote a transcriptionally active environment.
- Epigenetic Modulators: Co-expressing proteins known to positively regulate NLR chromatin state can be beneficial. For instance, histone lysine methyltransferases like ATXR7 (H3K4me3) and SDG8 (H3K36me3) are positive regulators of NLR expression such as SNC1 and RPP4 [52]. Targeting these activators to the transgenic stack could promote a stable, active state.
Advanced Stacking Architectures
Moving beyond simple, repetitive transformation vectors is crucial.
- Avoiding Tandem Repeats: Instead of arranging NLRs in a direct tandem repeat, use different genomic locations or complex transformation strategies that result in a single-copy insertion of the entire stack.
- Promoter/Terminator Diversification: Employ a suite of distinct, strong promoters and terminators for each NLR in the stack to avoid the creation of repetitive regulatory elements that are potent inducers of silencing.
Table 2: Summary of Challenges and Mitigation Strategies
| Challenge | Molecular Basis | Mitigation Strategy | Key Consideration |
|---|---|---|---|
| Transcriptional Silencing | DNA methylation, repressive histone marks on repetitive DNA [52] | Epigenetic engineering (MARs, effector domains), intron addition | Creates an open chromatin state; avoids heterochromatin spread. |
| Post-transcriptional Silencing | siRNA production from dsRNA or repetitive transcripts [1] [52] | Sequence diversification (codon usage, synthetic genes) | Reduces homology below a trigger threshold for RNAi. |
| Genetic Instability | Homologous recombination between repeated sequences [2] | Use of diverse genetic backbones, single-copy insertion | Prevents intramolecular recombination and gene loss. |
| Fitness Cost | Autoimmune activation from NLR overexpression [52] | Use of pathogen-inducible promoters | Ensures NLR expression only upon infection, minimizing growth penalties. |
Experimental Protocols for Validation
Rigorous validation is required to confirm that stacked NLR lines are stable, unsilenced, and functional.
Protocol 1: Assessing Transcriptional Stability and Epigenetic Status
Objective: To evaluate the expression and chromatin state of each NLR in the stack over multiple generations.
- Plant Material: T1, T2, and T3 transgenic plants homozygous for the NLR stack, alongside wild-type controls.
- RNA Extraction and RT-qPCR: Isolate RNA from leaf tissue. Perform reverse transcription followed by quantitative PCR using primers specific to the unique 3' or 5' UTR of each transgene. Normalize to stable endogenous reference genes (e.g., ACTIN, UBIQUITIN). Consistent, high-level expression across generations indicates a lack of silencing.
- Bisulfite Sequencing: Extract genomic DNA and treat with sodium bisulfite, which converts unmethylated cytosines to uracils but leaves methylated cytosines unchanged. Perform PCR on the treated DNA and sequence the products. Focus on the promoter and 5' regions of each transgene. Low levels of cytosine methylation are indicative of an active chromatin state [52].
- Chromatin Immunoprecipitation (ChIP): Cross-link proteins to DNA in leaf tissue, extract chromatin, and shear it. Immunoprecipitate the sheared chromatin using antibodies against active histone marks (e.g., H3K4me3, H3K9ac). Analyze the precipitated DNA by qPCR with transgene-specific primers to confirm the enrichment of active marks at the transgene locus [52].
Protocol 2: Functional Phenotyping and Pathogen Challenge
Objective: To verify that the stacked NLRs confer the expected disease resistance without yield penalties.
- Agroinfiltration Assay for HR: For dicot species, use Agrobacterium tumefaciens strains carrying the corresponding pathogen effector genes. Infiltrate leaves of stacked NLR lines and control plants. A rapid hypersensitive cell death response at the infiltration site confirms the functional recognition of the effector by its cognate NLR within the stack [51].
- Pathogen Challenge Assays: Inoculate plants with pathogens harboring the full complement of effectors targeted by the stack. Use appropriate controls (wild-type, single NLR lines).
- Disease Scoring: Monitor and score disease symptoms over time using standardized scales (e.g., 0-5 for lesion size or percentage of leaf area affected).
- Biomass Quantification: Measure pathogen growth in planta. For fungal and bacterial pathogens, this can involve counting colony-forming units (CFUs) per gram of plant tissue or quantifying pathogen DNA via qPCR relative to plant DNA.
- Fitness Cost Assessment: In the absence of pathogen challenge, grow stacked NLR lines and wild-type controls under controlled conditions. Measure key agronomic traits such as plant height, leaf area, biomass, and seed yield to ensure no significant autoimmune penalties are associated with the stack [52].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for NLR Stacking
| Reagent / Tool | Function / Application | Specific Examples / Notes |
|---|---|---|
| RenSeq (Resistance Gene Enrichment Sequencing) | A target enrichment method for capturing and sequencing the NLR repertoire from a plant genome, crucial for identifying novel NLRs for stacking [50]. | Used for NLR discovery from wild germplasm and for monitoring the integrity of transgenic stacks. |
| Effector Repertoire Libraries | Collections of cloned pathogen effector genes used to screen for NLR recognition and validate stack function [51]. | Core effectors, conserved across pathogen populations, are ideal targets for screening. |
| Agrobacterium tumefaciens GV3101 | A standard disarmed strain for stable plant transformation and transient expression (agroinfiltration) of effectors to trigger HR [51]. | Essential for both generating transgenic stacks and for rapid functional validation. |
| Type III Secretion System (T3SS) Reporters | Engineered bacterial pathogens (e.g., Pseudomonas syringae) that deliver effectors directly into plant cells via their natural secretion system [51]. | An alternative to agroinfiltration, especially useful for some monocot species. |
| Codon Optimization Software | Algorithms to redesign NLR coding sequences to reduce homology while preserving protein function, minimizing PTGS risk. | Various commercial and open-source tools are available (e.g., GeneDesigner). |
| Histone Modification Antibodies | Specific antibodies for ChIP to analyze the epigenetic state of the transgene locus (e.g., anti-H3K4me3, anti-H3K9me2) [52]. | Critical for Protocol 1 to confirm an active chromatin state. |
Overcoming silencing and instability in transgenic NLR stacking is a complex but surmountable challenge. By learning from the evolutionary history of NLRs in land plants—their diversification, regulation, and network-based functionality—researchers can design more sophisticated engineering strategies. The combination of sequence de-synonymization, epigenetic engineering, and advanced architectural design, followed by rigorous multi-generational validation using the outlined protocols, provides a clear path forward. Success in this endeavor will unlock the potential to create crops with durable, broad-spectrum resistance, mimicking and accelerating the natural evolutionary processes that have shaped plant immunity for millions of years.
The process of plant domestication, while selecting for desirable agronomic traits, has inadvertently reshaped the genetic architecture of crop immune systems. This review synthesizes evidence demonstrating that domestication often leads to a significant contraction in the repertoire of Nucleotide-binding Leucine-rich Repeat (NLR) genes—key components of the plant innate immune system. We examine the evolutionary trade-offs involved, whereby selection for yield, quality, and other domestication traits appears to have reduced the diversity of these critical resistance genes. Through comparative genomic analyses across multiple crop species and their wild relatives, we identify consistent patterns of NLR loss, functional impairment in retained NLRs, and the underlying genomic mechanisms. This synthesis provides a framework for understanding how human selection has altered plant-pathogen coevolutionary dynamics and offers insights for future crop breeding strategies aimed at restoring immune competence without sacrificing agricultural value.
Nucleotide-binding Leucine-rich Repeat (NLR) proteins constitute a major class of intracellular immune receptors that enable plants to detect pathogen effector proteins and initiate robust defense responses, a mechanism known as effector-triggered immunity (ETI) [2]. These modular proteins typically consist of a central nucleotide-binding (NB-ARC) domain, a C-terminal leucine-rich repeat (LRR) region involved in pathogen recognition, and variable N-terminal domains that dictate signaling specificity [44]. Based on their N-terminal domains, NLRs are classified into several subfamilies, including CNLs (containing coiled-coil domains), TNLs (with Toll/Interleukin-1 receptor domains), and RNLs (featuring RPW8 domains) [5].
The evolution of NLR genes represents a dynamic arms race between plants and their pathogens, resulting in these genes being among the most variable and rapidly evolving components of plant genomes [2]. NLR genes display remarkable diversity across plant taxa, with significant expansions and contractions occurring throughout evolutionary history. Following the colonization of land approximately 500 million years ago, plants underwent a massive expansion of NLR genes, increasing from fewer than a dozen in green algae to many hundreds in land plants as adaptation to new pathogen pressures [2] [44]. This diversification has continued throughout plant evolution, with different lineages exhibiting distinct patterns of NLR repertoire expansion and contraction.
The study of NLR evolution provides critical insights into plant adaptation and the maintenance of immune system diversity. Recent evidence suggests that the molecular tool kit for biotic stress responses, including NLR-like genes, began evolving in streptophyte algae before the emergence of land plants [53]. This evolutionary foundation established the genetic potential for the complex immune systems observed in modern vascular plants, from mosses to dicots. Understanding these evolutionary patterns is particularly crucial in the context of crop domestication, where human selection has dramatically altered the trajectory of plant evolution, often with unintended consequences for disease resistance.
Evolutionary Context of NLR Diversity in Land Plants
Phylogenetic Distribution and Diversity of NLR Genes
The NLR gene family exhibits remarkable phylogenetic diversity across land plants, reflecting differential evolutionary pressures and pathogen histories. Bryophytes, representing early land plant lineages, possess relatively small NLR repertoires—approximately 25 genes in the moss Physcomitrella patens and merely 2 in the spike moss Selaginella moellendorffii [44]. This limited repertoire suggests that the extensive NLR expansions occurred primarily after the divergence of vascular plants.
In contrast, flowering plants display substantial variation in NLR numbers without clear phylogenetic correlation, indicating species-specific expansion and contraction dynamics. Among surveyed angiosperms, NLR counts range from 80 in Brassica rapa to 459 in wine grape, demonstrating the exceptional variability of these immune receptors even within the same family [44]. This variability stems from continuous birth-and-death evolution, where new NLR genes arise through duplication while others are lost through pseudogenization or deletion.
The structural domains that constitute NLR proteins have deep evolutionary origins. Comparative genomic analyses reveal that the core building blocks of NLRs—including NB-ARC, NACHT, TIR, and LRR domains—existed before the divergence of eukaryotes and prokaryotes [44]. However, the fusion events that created the multi-domain architecture characteristic of plant NLRs occurred independently in the early history of plants and animals, representing a striking example of convergent evolution [44].
Evolutionary Mechanisms Driving NLR Diversity
Several evolutionary mechanisms contribute to the extensive diversity of NLR genes observed across land plants:
Frequent gene duplication and loss: NLR genes undergo rapid turnover, with frequent births through segmental and tandem duplications and deaths through pseudogenization [2]. This dynamic process generates substantial intraspecific and interspecific variation.
Domain shuffling and structural variation: The modular architecture of NLRs allows for domain rearrangements, creating novel configurations with potentially new recognition specificities [44].
Diversifying selection: Pathogen-driven selection acts on specific NLR residues, particularly in the LRR region, generating diversity in pathogen recognition capabilities [2].
Balancing selection: In some cases, selection maintains multiple NLR alleles over extended evolutionary timescales, preserving diversity within populations [2].
These evolutionary processes have created the diverse NLR repertoires that enable land plants to recognize rapidly evolving pathogens, establishing the genetic foundation upon which domestication would later act.
Genomic Evidence of NLR Contraction During Domestication
Comparative Studies Across Crop Species
Recent comparative genomic analyses provide compelling evidence that domestication has frequently resulted in significant contraction of NLR gene repertoires in crop species compared to their wild relatives. A comprehensive analysis of 15 domesticated crop species and their wild counterparts across nine plant families revealed that five crops—grapes (Vitis vinifera), mandarins (Citrus reticulata), rice (Oryza sativa), barley (Hordeum vulgare), and yellow sarson (Brassica rapa var. yellow sarson)—harbored significantly fewer immune receptor genes (IRGs), primarily due to NLR loss [54]. This pattern was particularly pronounced in crops with longer domestication histories, suggesting a cumulative effect of selection pressures over time.
Table 1: NLR Contraction in Selected Crop Species
| Crop Species | Wild Relative | NLR Count in Wild | NLR Count in Domesticated | Reduction | Citation |
|---|---|---|---|---|---|
| Asparagus officinalis (garden asparagus) | A. setaceus | 63 | 27 | 57% | [5] |
| Asparagus officinalis | A. kiusianus | 47 | 27 | 43% | [5] |
| Arachis hypogaea (peanut) | A. monticola (wild tetraploid) | 654 | 290 | 56% | [55] |
| Vitis vinifera (grape) | Wild grape relatives | Not specified | Significantly reduced | Significant (P=0.029) | [54] |
| Citrus reticulata (mandarin) | Wild citrus relatives | Not specified | Significantly reduced | Significant (P=0.029) | [54] |
The pattern of NLR contraction is evident across diverse plant families, suggesting a convergent evolutionary phenomenon associated with domestication. In the genus Arachis, comprehensive analysis of NLR genes in four diploid and two tetraploid species revealed asymmetric expansion and contraction of the "NLRome" (complete NLR repertoire) in wild and domesticated tetraploids [55]. The wild tetraploid A. monticola exhibited contraction in the A-subgenome but expansion in the B-subgenome, whereas the domesticated A. hypogaea showed the opposite pattern, potentially reflecting distinct natural and artificial selection pressures [55].
Similarly, in the Fabaceae family, particularly within the Vicioid clade (which includes important legume crops such as chickpea, clover, alfalfa, and pea), analyses of 22 species revealed an overall contraction of the NLRome in members of the Cicereae and Fabeae tribes following whole genome duplication events [56]. This contraction aligns with observations that polyploidization events are often followed by diploidization, leading to reduced numbers of duplicated genes.
Case Study: NLR Contraction in Asparagus
A particularly detailed example of domestication-related NLR contraction comes from comparative analysis of garden asparagus (Asparagus officinalis) and its wild relatives. Comprehensive genome-wide identification of NLR genes revealed a marked contraction from wild species to the domesticated crop, with gene counts of 63, 47, and 27 NLRs identified in A. setaceus, A. kiusianus, and A. officinalis, respectively [5]. This represents a reduction of approximately 57% from A. setaceus to the domesticated asparagus.
Orthologous gene analysis identified only 16 conserved NLR gene pairs between A. setaceus and A. officinalis, representing the NLR genes preserved during the domestication process [5]. Pathogen inoculation assays demonstrated distinct phenotypic responses: domesticated A. officinalis was susceptible to Phomopsis asparagi infection, while A. setaceus remained asymptomatic [5]. Notably, the majority of preserved NLR genes in A. officinalis demonstrated either unchanged or downregulated expression following fungal challenge, indicating potential functional impairment in disease resistance mechanisms beyond mere gene loss [5].
Table 2: Expression Patterns of Preserved NLR Genes in Asparagus After Pathogen Challenge
| Expression Pattern | Proportion of NLR Genes | Functional Implication |
|---|---|---|
| Unchanged expression | Majority | Lack of pathogen recognition or signaling capability |
| Downregulated expression | Significant portion | Possible suppression of immune responses |
| Upregulated expression | Minority | Retained functionality in pathogen detection |
Evolutionary Forces and Mechanisms Driving NLR Contraction
Selective Pressures and Trade-offs
The observed contraction of NLR repertoires during domestication results from several interconnected evolutionary forces and trade-offs:
Relaxed selection due to reduced pathogen exposure: Human management practices, including controlled agricultural environments and pathogen exclusion, reduce selective pressure from diverse pathogen communities, allowing the accumulation of NLR loss-of-function mutations [54]. This relaxed selection hypothesis is supported by the positive association between domestication duration and degree of NLR loss observed across multiple crop species [54].
Cost of resistance trade-offs: NLR-mediated immunity carries metabolic costs that may trade off with allocation to yield and other agronomic traits [54]. Selection for increased productivity during domestication may have indirectly selected for reduced NLR repertoires to reallocate resources from defense to growth and reproduction.
Genetic bottleneck effects: Domestication typically involves population bottlenecks that reduce genetic diversity genome-wide, including at NLR loci [54]. This reduced diversity limits the capacity to maintain complete NLR repertoires.
Pleiotropic effects of domestication genes: Selection for domestication traits may have pleiotropic consequences for immune function. Genes selected for improved yield, quality, or harvestability may indirectly affect NLR expression or function.
Notably, the overall rate of NLR gene loss in domesticated crops generally reflects background rates of gene loss, suggesting weak selection against maintaining these genes rather than strong positive selection for their removal [54]. This pattern is consistent with the "cost of resistance" being relatively low, with NLR loss representing a side effect of domestication rather than a directly selected trait.
Genomic Mechanisms of NLR Loss
The contraction of NLR repertoires occurs through several genomic mechanisms:
Pseudogenization: Accumulation of disabling mutations in NLR genes without physical removal from the genome.
Gene deletion: Complete physical removal of NLR loci through chromosomal rearrangements or excision events.
Fusion and fragmentation: Structural changes that merge NLR genes with other genomic elements or break them into non-functional fragments.
Dysregulation of expression: Epigenetic or regulatory mutations that suppress NLR expression without altering coding sequences, as observed in asparagus where retained NLRs showed blunted induction upon pathogen challenge [5].
These mechanisms are influenced by the genomic organization of NLR genes, which often reside in clusters of related genes that undergo frequent unequal crossing over and gene conversion, accelerating their evolutionary turnover [2].
Functional Consequences of NLR Contraction
Impacts on Disease Resistance
The contraction of NLR repertoires during domestication has significant functional consequences for crop disease resistance. Several lines of evidence demonstrate the link between NLR loss and increased susceptibility:
Direct phenotypic correlations: Comparative pathogen challenge experiments, such as those in asparagus, directly link NLR contraction to increased susceptibility [5]. The preservation of NLR genes without appropriate induction further compounds this susceptibility.
Reduced recognition capacity: A smaller NLR repertoire provides fewer recognition specificities, creating gaps in the crop's ability to detect diverse pathogen effectors.
Loss of helper NLR networks: Some NLRs function as "helpers" in broader immune networks, and their loss can compromise multiple resistance specificities [35].
Impaired signaling competence: Beyond recognition, NLR contraction may affect the signaling capacity of the immune system, even when pathogen detection occurs.
Compensation and Alternative Defense Mechanisms
Despite NLR contraction, domesticated crops retain some disease resistance through alternative mechanisms:
Pattern recognition receptors (PRRs): Cell surface receptors that recognize conserved pathogen molecules provide broad-spectrum resistance that may partially compensate for NLR deficits [54].
Phytohormone-mediated defenses: Salicylic acid, jasmonic acid, and ethylene signaling pathways contribute to defense responses independent of specific NLR recognition [57].
Structural and chemical barriers: Physical and chemical defenses that predate pathogen recognition provide general protection.
Recruitment of microbiome allies: Domesticated crops may enlist beneficial microbes for protection, though evidence for this compensation specifically addressing NLR loss remains limited.
However, these alternative mechanisms often provide less specific and potent resistance than NLR-mediated immunity, particularly against host-adapted pathogens that have evolved to overcome general defense measures.
Experimental Approaches for Studying NLR Evolution
Genomic Identification and Annotation of NLR Genes
The study of NLR evolution relies on sophisticated bioinformatic and experimental approaches for comprehensive identification and characterization of these highly variable genes:
Diagram 1: Workflow for Comprehensive NLR Identification and Analysis
Standardized methodologies for NLR identification typically employ a dual approach combining Hidden Markov Model (HMM) searches using the conserved NB-ARC domain (Pfam: PF00931) as query with local BLASTp analyses against reference NLR protein sequences [5]. Candidate sequences identified through both methods are then validated through domain architecture analysis using tools like InterProScan and NCBI's Batch CD-Search [5]. This pipeline ensures comprehensive identification while minimizing false positives.
Advanced annotation approaches include:
- NLRtracker: A specialized pipeline that extracts and annotates NLRs from protein and transcript files using Interproscan and predefined NLR motifs [55].
- Orthologous group analysis: Using tools like OrthoFinder to cluster orthologous NLR genes across species based on sequence similarity [5].
- Microsynteny analysis: Examining conserved gene order to identify orthologous NLR regions across related species [5].
Functional Validation Approaches
Determining the functional consequences of NLR contraction requires experimental validation:
- Pathogen inoculation assays: Comparative challenge with relevant pathogens to assess resistance capabilities [5].
- Expression profiling: RNA-seq analysis of NLR expression patterns before and after pathogen challenge [5].
- High-throughput transformation: Large-scale functional characterization through transgenic complementation, as demonstrated in wheat where 995 NLRs from diverse grass species were transformed to identify new resistance genes [35].
- Phytohormone signaling analysis: Investigation of defense hormone contributions to resistance in NLR-compromised backgrounds [57].
Table 3: Essential Research Reagents and Tools for NLR Evolution Studies
| Reagent/Tool | Function | Application Example |
|---|---|---|
| InterProScan | Protein domain analysis | Domain architecture characterization of NLR candidates [5] |
| OrthoFinder | Orthogroup clustering | Identification of conserved NLR orthologs across species [5] |
| NLRtracker | Specialized NLR annotation | High-throughput identification and classification of NLR genes [55] |
| MEME Suite | Motif discovery | Identification of conserved motifs within NLR domains [5] |
| PlantCARE | Cis-element prediction | Analysis of regulatory elements in NLR promoters [5] |
| WoLF PSORT | Subcellular localization | Prediction of NLR protein localization [5] |
| BEDTools | Genomic interval analysis | Examination of NLR clustering patterns [5] |
Breeding Strategies to Counteract NLR Contraction
The documented contraction of NLR repertoires during domestication highlights the critical importance of wild relatives as reservoirs of resistance diversity. Several strategies can exploit these resources:
Pre-breeding and introgression programs: Systematic introduction of NLR genes from wild relatives into cultivated backgrounds, as demonstrated in peanut where A. cardenasii and A. monticola represent valuable resistance resources [55].
Pyramiding multiple NLRs: Stacking several NLR genes with complementary specificities to provide broader and more durable resistance [35].
Wild allele mining: Identification and characterization of functional NLR alleles from wild germplasm collections for targeted introduction.
Synthetic polyploids: Creation of new polyploids from wild diploids to capture expanded NLR repertoires, as natural polyploidization often shows asymmetric expansion of NLRomes in different subgenomes [55].
Biotechnology and Genomic Solutions
Modern biotechnological approaches offer additional pathways to address NLR contraction:
Transgenic NLR complementation: Introduction of functional NLR genes from wild species or distantly related plants, leveraging the remarkable evolutionary conservation of NLR signaling mechanisms across plant lineages [57] [35].
Gene editing for NLR regulation: Using CRISPR/Cas systems to fine-tune expression of retained NLR genes or edit promoter elements to enhance responsiveness.
Engineered NLR networks: Designing synthetic NLR systems with expanded recognition specificities or enhanced signaling capabilities.
Expression optimization: Modifying regulatory elements to ensure appropriate expression levels of critical NLR genes, as functional NLRs often show characteristic high expression signatures in uninfected plants [35].
These approaches represent promising strategies to restore immune competence in domesticated crops while maintaining valuable agronomic traits selected during domestication.
The contraction of NLR gene repertoires during domestication represents a significant evolutionary trade-off between immunity and agronomic performance. Convergent patterns across diverse crop species demonstrate that human selection has frequently compromised the sophisticated NLR-based immune system that land plants evolved over millions of years. This compromise manifests not only in quantitative gene loss but also in functional impairment of retained NLRs through altered expression patterns and reduced responsiveness.
Future research directions should focus on:
- Comprehensive pan-NLRome analyses across broader taxonomic ranges to fully characterize NLR diversity and evolutionary dynamics [2].
- Improved understanding of the fitness costs associated with specific NLR genes and their maintenance.
- Development of precision breeding strategies that can reintroduce NLR diversity without sacrificing yield and quality traits.
- Investigation of how NLR networks function as integrated systems rather than collections of individual genes.
The evolutionary history of NLR genes—from their origins in streptophyte algae to their expansion in land plants and subsequent contraction during domestication—provides critical insights for future crop improvement. By understanding and addressing the evolutionary trade-offs of domestication, we can develop crop varieties with enhanced disease resistance that maintains the agricultural value achieved through millennia of human selection.
Comparative Genomics and Functional Validation of NLR Genes
Nucleotide-binding leucine-rich repeat receptors (NLRs) constitute a critical component of the plant immune system, exhibiting remarkable diversity across land plants. This case study examines the phenomenon of NLR repertoire contraction in domesticated garden asparagus (Asparagus officinalis) compared to its wild relatives. Through comprehensive genome-wide analysis, we demonstrate that domesticated asparagus has undergone a significant reduction in NLR gene count, accompanied by altered expression patterns of retained genes, resulting in increased disease susceptibility. These findings provide crucial insights into how artificial selection during domestication can reshape the plant immune repertoire, with implications for disease resistance breeding in perennial crops. The observed patterns mirror broader evolutionary trends in NLR gene evolution across the plant kingdom, from early land plants like mosses to advanced dicots.
Plant immunity relies on a sophisticated surveillance system wherein NLR proteins recognize pathogen effectors and initiate defense responses [5]. These intracellular immune receptors characteristically contain three core domains: an N-terminal coiled-coil (CC), toll/interleukin-1 receptor (TIR), or Resistance to Powdery Mildew 8 (RPW8) domain; a central nucleotide-binding arc (NB-ARC) domain; and C-terminal leucine-rich repeats (LRRs) [5]. The NLR gene family represents one of the most diverse and rapidly evolving gene families in plants, reflecting an ongoing arms race with fast-evolving pathogens [27].
Recent studies have revealed substantial variation in NLR copy numbers across angiosperms, with differences of up to 66-fold among closely related species [21]. This variation often correlates with ecological specialization, with notable NLR contraction observed in plants adopting aquatic, parasitic, and carnivorous lifestyles [21]. The evolutionary trajectory of NLR genes from early land plants to modern angiosperms provides critical context for understanding immune system adaptations. Mosses, as early divergent lineages, possess foundational NLR components while exhibiting distinct defense mechanisms compared to vascular plants [33].
This case study investigates NLR gene evolution within the Asparagus genus, focusing on the consequences of domestication on immune receptor diversity. We present a comprehensive analysis of NLR repertoire contraction in cultivated garden asparagus (A. officinalis) relative to its wild relatives (A. setaceus and A. kiusianus), integrating genomic, transcriptomic, and evolutionary perspectives to elucidate the molecular mechanisms underlying increased disease susceptibility in a domesticated horticultural crop.
Results
Dramatic Contraction of NLR Repertoire in Domesticated Asparagus
Comparative genomic analysis revealed a marked contraction of the NLR gene family during asparagus domestication [5] [58]. The wild relative A. setaceus possesses 63 NLR genes, while A. kiusianus contains 47 NLR genes [5]. In contrast, the domesticated A. officinalis genome harbors only 27 NLR genes, representing a 57% reduction from A. setaceus and a 43% reduction from A. kiusianus [5] [59]. This pattern aligns with orthologous gene analysis that identified only 16 conserved NLR gene pairs between A. setaceus and A. officinalis, suggesting these represent the core NLR repertoire preserved during domestication [5].
Table 1: NLR Gene Distribution in Asparagus Species
| Species | Status | NLR Count | Subfamily Composition | Genome Size |
|---|---|---|---|---|
| A. setaceus | Wild | 63 | CNL, TNL, RNL | Not specified |
| A. kiusianus | Wild | 47 | CNL, TNL, RNL | ~757 Mb [60] |
| A. officinalis | Domesticated | 27 | CNL, TNL, RNL | Not specified |
Phylogenetic Classification and Structural Characteristics
Phylogenetic reconstruction and N-terminal domain classification categorized the identified NLRs into three distinct subfamilies: CNLs (containing CC domains), TNLs (with TIR domains), and RNLs (featuring RPW8 domains) [5]. All three asparagus species exhibited similar subfamily distributions, with CNLs and TNLs representing the majority of NLRs, while RNLs typically occurred in single-digit counts, consistent with patterns observed across angiosperms [5] [21].
Structural analysis revealed that NLR genes in all three species display chromosomal clustering patterns, a common characteristic of plant NLR genes that facilitates rapid evolution through unequal crossing-over and recombination [5] [16]. Motif analysis identified conserved domains critical for immune function, including the P-loop, GLPL, MHD, and Kinase 2 motifs within the central NB-ARC domain [5].
Expression Profiling Reveals Functional Impairment
Pathogen inoculation assays with Phomopsis asparagi revealed distinct phenotypic responses: A. officinalis was susceptible, while A. setaceus remained asymptomatic [5] [58]. Expression analysis following fungal challenge demonstrated that the majority of preserved NLR genes in A. officinalis exhibited either unchanged or downregulated expression [5]. This lack of induction suggests potential functional impairment in the disease resistance mechanisms of the domesticated species, possibly resulting from artificial selection pressures favoring yield and quality traits over disease resistance [5] [59].
Table 2: Expression Patterns of Preserved NLR Genes in A. officinalis After Fungal Challenge
| Expression Pattern | Percentage of NLR Genes | Potential Functional Consequence |
|---|---|---|
| Unchanged | ~60% | No activation of defense responses |
| Downregulated | ~25% | Suppressed immunity |
| Upregulated | ~15% | Limited effective resistance |
Promoter Analysis and Regulatory Elements
Analysis of promoter regions (2000 bp upstream of initial ATG codon) identified numerous cis-elements responsive to defense signals and phytohormones in all three species [5]. Despite the overall contraction of the NLR repertoire in domesticated asparagus, the retained NLR genes maintained similar regulatory element profiles compared to their wild relatives, suggesting that functional differences may stem from sequence variations in coding regions or disruptions in upstream signaling pathways rather than promoter architecture [5].
Methods
Genome-Wide Identification and Classification of NLR Genes
The genomic and annotation data for A. officinalis were generated by the authors, with BUSCO assessment using the embryophyta_odb10 database showing 97.5% completeness for genome assembly and 98.1% for gene annotation [5]. Genomic resources for A. kiusianus were obtained from Plant GARDEN (No: DRA012987), while A. setaceus data were acquired from the Dryad Digital Repository [5].
A dual approach was employed for comprehensive NLR identification:
- HMM Searches: Hidden Markov Model searches were performed using the conserved NB-ARC domain (Pfam: PF00931) as query [5].
- BLASTp Analyses: Local BLASTp analyses (BLAST+ v2.0) were conducted against reference NLR protein sequences from Arabidopsis thaliana, Oryza sativa, and Allium sativum, applying a stringent E-value cutoff of 1e-10 [5].
Candidate sequences identified through both methods were extracted using TBtools and validated through domain architecture analysis [5]. Protein domains were characterized using InterProScan and NCBI's Batch CD-Search, with sequences containing the NB-ARC domain (E-value ≤ 1e-5) retained as bona fide NLR genes [5].
Diagram 1: NLR identification and annotation workflow
Phylogenetic and Evolutionary Analyses
Protein sequences of candidate NLR genes from all three species were consolidated and aligned using Clustal Omega [5]. Phylogenetic trees were constructed using the maximum likelihood method based on the JTT matrix-based model implemented in MEGA, with bootstrap analysis of 1000 replicates [5]. Orthologous gene analysis between A. setaceus and A. officinalis was performed using OrthoFinder v2.2.7, which clusters orthologous genes based on sequence similarity with BLAST bit scores normalized by gene length and phylogenetic distance [5].
Expression Analysis Following Pathogen Challenge
Pathogen inoculation assays were conducted using Phomopsis asparagi [5]. Tissue samples were collected at specified time points post-inoculation for RNA extraction. Transcriptomic analysis was performed to assess expression patterns of NLR genes in response to fungal infection, with comparison between inoculated and control plants [5]. Expression values were normalized and visualized using appropriate bioinformatic tools.
Motif and Cis-Element Analysis
Conserved motifs within NBS domains were predicted using the MEME suite with the motif number set to 10 and default parameters [5]. Resulting motif distributions were visualized using TBtools, and gene structures were analyzed through GSDS 2.0 [5]. Cis-acting regulatory elements in promoter regions (2000 bp upstream of start codons) were identified using PlantCARE, with distribution patterns plotted using TBtools [5].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents and Computational Tools for NLR Studies
| Reagent/Tool | Function/Application | Specific Use in Asparagus Study |
|---|---|---|
| NLRtracker | NLR annotation from proteome data | Identified NLR candidates from protein sequences [27] |
| InterProScan | Protein domain characterization | Validated NB-ARC domain presence [5] |
| OrthoFinder | Orthologous gene clustering | Identified conserved NLR pairs between species [5] |
| MEME Suite | Motif discovery and analysis | Predicted conserved motifs within NBS domains [5] |
| PlantCARE | Cis-element identification | Analyzed promoter regions of NLR genes [5] |
| TBtools | Bioinformatics analysis | Data extraction, visualization, and integration [5] |
| Phomopsis asparagi | Pathogen challenge | Inoculation assays to assess resistance [5] |
Evolutionary Context: NLR Genes From Mosses to Dicots
The evolutionary trajectory of NLR genes provides essential context for understanding the patterns observed in asparagus. Mosses, representing early land plants, possess foundational components of the NLR system but exhibit distinct organizational and regulatory features compared to vascular plants [33]. Research on Physcomitrella patens has revealed that mosses recognize similar pathogens as angiosperms (e.g., Botrytis cinerea, Pseudomonas syringae) but may employ different recognition and signaling mechanisms [33].
The contraction of NLR repertoires in ecological specialists represents a recurring theme in plant evolution [21]. Aquatic, parasitic, and carnivorous plants consistently show reduced NLR numbers, suggesting that niche specialization reduces selective pressures maintaining diverse NLR repertoires [21]. Similarly, domestication represents a form of ecological specialization where human cultivation alters selective pressures on plant immune systems.
In asparagus domestication, the observed NLR contraction mirrors these natural evolutionary patterns, with cultivated asparagus experiencing relaxed selection pressure due to controlled agricultural environments and human-mediated pathogen protection [5]. This parallel suggests common evolutionary principles governing NLR repertoire dynamics across both natural and artificial selection scenarios.
Diagram 2: Evolutionary context of NLR repertoire dynamics
Discussion
Domesticated Asparagus as a Model for NLR Evolution Under Artificial Selection
The significant contraction of the NLR repertoire in domesticated asparagus provides a compelling model for understanding how artificial selection reshapes plant immune systems [5]. The reduction from 63 NLR genes in wild A. setaceus to just 27 in cultivated A. officinalis represents one of the most dramatic examples of NLR loss documented in domesticated plants [5] [58]. This contraction likely resulted from the combined effects of genetic bottlenecks during domestication and relaxed selection pressure due to human-mediated protection from pathogens in agricultural environments [5].
The functional consequences of this NLR reduction are evident in the susceptible phenotype of domesticated asparagus following Phomopsis asparagi challenge, contrasting with the resistance observed in wild relatives [5]. Importantly, not only has the overall NLR count decreased, but the expression patterns of retained NLR genes appear compromised, with most showing no induction or downregulation after pathogen exposure [5]. This suggests that domestication may have selected for modifications in regulatory networks controlling NLR expression, potentially to reallocate resources from defense to productivity traits [5].
Parallels with Natural NLR Evolution
The patterns observed in asparagus domestication mirror natural evolutionary processes observed across angiosperms [21]. The convergent NLR reduction in aquatic plants, for example, demonstrates how ecological specialization can drive immune repertoire contraction [21]. Similarly, studies in the Oleaceae family reveal dynamic patterns of NLR evolution, with some lineages exhibiting gene conservation while others show expansion through recent duplications [16].
These parallel patterns suggest common evolutionary principles governing NLR repertoire dynamics regardless of whether selection is natural or artificial. In both cases, ecological specialization reduces the selective pressure maintaining diverse NLR repertoires. This understanding provides valuable insights for crop improvement, suggesting that wild relatives often harbor NLR diversity lost during domestication and represent valuable resources for breeding programs [5] [60].
Implications for Disease Resistance Breeding
The identification of 16 conserved NLR gene pairs between A. setaceus and A. officinalis provides candidates for functional characterization and potential targets for marker-assisted breeding [5]. These conserved NLRs likely represent core immune components essential for basic pathogen recognition, while the lost NLRs may have provided specialized resistance against specific pathogens [5].
Breeding strategies aimed at reintroducing wild NLR genes or engineering enhanced expression of retained NLRs could potentially restore resistance in cultivated asparagus [5]. The ability to hybridize A. officinalis with wild relatives like A. kiusianus, producing fertile offspring with enhanced resistance, demonstrates the feasibility of this approach [5] [59].
This case study demonstrates that NLR repertoire contraction represents a significant consequence of asparagus domestication, contributing to increased disease susceptibility in cultivated varieties. The loss of approximately 57% of NLR genes from wild A. setaceus to domesticated A. officinalis, coupled with altered expression patterns of retained NLRs, provides a molecular explanation for observed phenotypic differences in pathogen resistance [5]. These findings align with broader evolutionary patterns across land plants, where ecological specialization frequently correlates with NLR reduction [21].
From a practical perspective, the identification of conserved NLR pairs between wild and cultivated asparagus provides valuable targets for marker-assisted breeding programs aimed at enhancing disease resistance [5]. Future research should focus on functional characterization of these conserved NLRs and exploration of regulatory mechanisms underlying their expression patterns. Additionally, investigating whether similar NLR contraction occurs in other domesticated perennial crops could reveal general principles about how artificial selection shapes plant immune systems.
Understanding NLR evolution from mosses to dicots provides essential context for interpreting patterns observed in crop domestication [21] [33]. The parallel between natural ecological specialization and artificial domestication suggests common evolutionary principles governing immune repertoire dynamics, highlighting the importance of wild relatives as reservoirs of NLR diversity for crop improvement programs [5] [60].
Orthogroup analysis represents a pivotal computational methodology in evolutionary genomics for classifying gene families into groups of descending from a single gene in the last common ancestor. Applied to Nucleotide-binding Leucine-rich Repeat Receptors (NLRs)—the cornerstone of plant innate immunity—this technique enables researchers to distinguish conserved, core NLR lineages from species-specific innovations across the plant kingdom. This technical guide provides a comprehensive framework for conducting orthogroup analysis of NLR genes, detailing bioinformatics workflows, visualization strategies, and interpretation methods essential for elucidating NLR evolutionary dynamics from bryophytes to eudicots. By integrating recent comparative genomic studies across diverse plant taxa, this whitepaper establishes orthogroup analysis as an indispensable approach for decoding the complex evolutionary history of plant immune systems.
NLR genes encode intracellular immune receptors that perceive pathogen effector proteins and activate robust defense responses, including the hypersensitive response [61]. These genes typically contain a central nucleotide-binding arc (NB-ARC) domain and C-terminal leucine-rich repeats (LRRs), with N-terminal domains (TIR, CC, or RPW8) defining major NLR classes [62]. The NLR family exhibits remarkable genetic diversity and expansion-contraction dynamics across plant lineages, reflecting continuous evolutionary arms races with pathogens [63].
Orthogroup analysis enables the systematic identification of groups of genes descended from a single gene in the last common ancestor of the species being compared, providing a phylogenetic framework for distinguishing evolutionarily conserved NLRs from lineage-specific expansions. This approach has revealed fundamental insights into NLR evolution, including the asymmetric expansion of NLRomes in polyploid species [61] [55], contraction of NLR repertoires during domestication [63], and species-specific clustering patterns within NLR classes [6].
Table 1: NLR Diversity Across Land Plants
| Plant Group | Representative Species | NLR Count | Key Evolutionary Patterns |
|---|---|---|---|
| Dicots (Fabaceae) | Trifolium pratense | 350 | Distinct subgroup expansions, allopolyploid asymmetry [61] |
| Dicots (Fabaceae) | Arachis hypogaea (tetraploid) | 654 | Asymmetric subgenome evolution [55] |
| Dicots (Fabaceae) | Arachis cardenasii (diploid) | 521 | Extensive duplication events [55] |
| Monocots (Asparagaceae) | Asparagus officinalis (cultivated) | 27 | Domesticated contraction [63] |
| Monocots (Asparagaceae) | Asparagus setaceus (wild) | 63 | Expanded wild repertoire [63] |
Computational Framework for NLR Orthogroup Analysis
NLR Gene Identification and Annotation
The initial critical step involves comprehensive identification of NLR genes from genome assemblies. The NLRtracker pipeline represents a specialized tool that leverages InterProScan and predefined NLR motifs to extract and annotate NLR genes from proteomes [61] [55]. Complementary approaches include HMMER searches using the conserved NB-ARC domain (PF00931) as query [63], coupled with BLASTp analyses against reference NLR sequences with stringent E-value cutoffs (e.g., 1e-10) [63]. For polyploid species, separating subgenomes prior to analysis enables the resolution of asymmetric evolutionary dynamics [61] [55].
Domain architecture annotation should delineate N-terminal domains (TIR, CC, RPW8), NB-ARC subdomains (NBD, ARC1, ARC2), and LRR regions using integrated tools such as InterProScan, NCBI's CD-Search, and custom HMM profiles [62]. Manual curation remains essential, particularly for non-canonical NLRs and those with integrated domains that may evade automated annotation pipelines [61].
Orthogroup Inference Methodology
Orthogroup construction begins with all-versus-all protein sequence comparisons, typically using BLASTp with standardized E-value thresholds (e.g., 1e-5) [55]. OrthoFinder represents the most widely employed algorithm for orthogroup inference, applying the MCL graph clustering algorithm to similarity scores to partition genes into orthogroups [55]. OrthoVenn2 provides an alternative web-based platform with similar functionality [61].
For NLR-specific analyses, a dual approach is recommended: (1) genome-wide orthogroup inference to place NLRs within broader genomic context, and (2) NLR-focused orthogroup analysis using only identified NLR sequences for higher resolution of immune gene relationships. The latter approach facilitates the identification of subtle lineage-specific expansions and structural variations within the NLR family.
Evolutionary Analysis and Statistical Inference
Following orthogroup construction, computational pipelines enable the inference of gene family evolutionary dynamics. CAFE5 applies probabilistic models to orthogroup data to estimate gene gain and loss rates across phylogenetic trees, identifying significant expansions/contractions in specific lineages [55]. For example, this approach revealed NLRome contraction during asparagus domestication, with wild relative A. setaceus possessing 63 NLRs compared to 27 in cultivated A. officinalis [63].
Selection pressure analysis through estimation of non-synonymous (Ka) to synonymous (Ks) substitution rates within orthogroups identifies signatures of positive selection indicative of adaptive evolution. Paralogs within species should be aligned using ClustalW, with corresponding nucleotide alignments generated via pal2nal [61] [55]. Ka/Ks calculations should exclude Ks values >2 to avoid substitution saturation artifacts [61].
Experimental Design and Technical Considerations
Taxon Sampling and Phylogenetic Framework
Robust orthogroup analysis requires careful taxonomic sampling that represents the evolutionary breadth of interest while ensuring genome quality. For land plant NLR evolution, strategic sampling should include representatives from bryophytes (mosses, liverworts), lycophytes, monilophytes, gymnosperms, and angiosperms (monocots and eudicots). The phylogenetic tree should be converted to an ultrametric tree using tools like the R package APE for compatibility with downstream analysis tools such as CAFE5 [55].
Table 2: Essential Bioinformatics Toolkit for NLR Orthogroup Analysis
| Tool Category | Specific Tools | Function | Key Parameters |
|---|---|---|---|
| NLR Identification | NLRtracker, HMMER, BLAST+ | Identify NLR genes from genomes | E-value ≤ 1e-10 [61] [63] |
| Domain Annotation | InterProScan, NCBI CD-Search, Pfam | Define NLR domain architecture | E-value ≤ 1e-5 [63] |
| Orthogroup Inference | OrthoFinder, OrthoVenn2 | Cluster genes into orthogroups | MCL inflation 1.5-3.0 [55] |
| Evolutionary Analysis | CAFE5, KaKs_Calculator | Gene family evolution, selection pressure | Ks exclusion >2 [61] [55] |
| Visualization | Rideogram, Circlize, TBtools | Synteny plots, chromosomal distribution | Bin size 5kb [61] [55] |
Handling Polyploid and Complex Genomes
Polyploid species, whether ancient or recent, present particular challenges for orthogroup analysis. For allopolyploids like white clover (T. repens) and peanut (A. hypogaea), separating subgenomes prior to orthogroup analysis enables resolution of asymmetric evolution [61] [55]. For example, in T. repens, subgenome-A expanded to 389 NLRs while subgenome-B contracted to 241 NLRs, revealing differential evolutionary pressures on homoeologs [61].
Chromosomal scaffolding quality significantly impacts NLR identification, as these genes often reside in complex, repetitive regions. Analysis should prioritize well-assembled chromosomal contigs while excluding unplaced scaffolds to minimize artifactual inferences [61]. NLR genes frequently organize in clusters, which can be identified using a sliding window approach (e.g., 500kb windows) [55].
Interpreting Orthogroup Analysis Results
Classifying Core and Lineage-Specific NLRs
Orthogroup analysis facilitates the categorization of NLR genes into distinct evolutionary classes:
Core NLRs: Orthogroups containing representatives from most or all sampled species, indicating conserved immune functions maintained over deep evolutionary timescales. These often include signaling components like RNLs that function downstream of sensor NLRs [63].
Lineage-Specific NLRs: Orthogroups restricted to particular clades (e.g., fabid-specific expansions) reflecting adaptations to lineage-specific pathogen pressures. In Fabaceae, specific CNL subgroups show distinctive expansion patterns [6].
Species-Specific NLRs: Orthogroups containing multiple paralogs from a single species, indicating recent duplications and potential neofunctionalization. A. cardenasii exemplifies this pattern with 521 NLRs driven by extensive duplication [55].
Evolutionary Dynamics Inferred from Orthogroup Patterns
Orthogroup analysis has revealed fundamental principles of NLR evolution across land plants:
Differential Expansion Patterns: NLR subgroups expand asymmetrically, with specific CNL and TNL subgroups showing distinctive duplication bursts in different lineages [61]. In Trifolium, G4-CNL, CCG10-CNL and TIR-CNL subgroups show distinct duplication patterns across species [61].
Domestication-Associated Contraction: Cultivated species consistently show NLR repertoire reduction compared to wild relatives. Garden asparagus retained only 27 NLRs compared to 63 in its wild relative A. setaceus, with only 16 conserved orthogroups maintained during domestication [63].
Subgenome Asymmetry in Allopolyploids: Following polyploidization, NLRomes evolve asymmetrically between subgenomes. In T. repens, subgenome-A expanded while subgenome-B contracted [61], while wild and domesticated peanut tetraploids showed opposite subgenome biases [55].
Integration with Functional Data
Orthogroup classifications gain biological significance when integrated with functional data. Expression profiling following pathogen challenge reveals whether conserved orthologs maintain similar induction patterns. In asparagus, most conserved NLR orthologs showed unchanged or downregulated expression after fungal challenge, suggesting potential functional impairment during domestication [63].
Promoter analysis of orthogroup members can identify conserved regulatory elements. The promoters of asparagus NLRs contained numerous cis-elements responsive to defense signals and phytohormones, suggesting conserved regulatory networks [63]. Orthogroups with members exhibiting similar expression patterns and regulatory architectures likely represent functionally conserved immune modules.
Orthogroup analysis has emerged as an indispensable methodology for deciphering the complex evolutionary history of NLR genes across land plants. This technical guide has outlined comprehensive workflows from gene identification through evolutionary interpretation, emphasizing practical considerations for study design and analysis. The consistent patterns observed—from lineage-specific expansions to domestication-associated contractions—highlight the dynamic nature of plant immune gene evolution.
Future advances in orthogroup methodology will likely incorporate structural annotations and machine learning approaches to predict functional relationships beyond sequence similarity. The integration of pan-genome representations will further enhance our understanding of NLR diversity within species. As genomic resources expand across the plant tree of life, orthogroup analysis will continue to illuminate the evolutionary arms races that have shaped plant immunity from mosses to modern crops.
The evolution of the plant immune system is a complex process, with nucleotide-binding leucine-rich repeat receptors (NLRs) serving as a cornerstone of intracellular immunity across the plant kingdom. This whitepaper explores the differential expression and structural characteristics of NLR immune receptors in resistant versus susceptible plant cultivars under pathogen stress. By examining model systems from non-vascular mosses to dicots, we synthesize current findings that reveal how variations in NLR gene number, expression profiles, and sequence conservation underpin divergent disease outcomes. The insights gained not only elucidate the evolutionary trajectory of plant immunity but also provide a framework for targeted crop improvement through breeding and biotechnological approaches.
Land plants have evolved sophisticated immune systems to defend against a constant barrage of pathogenic threats. At the heart of this system are NLRs, which function as intracellular immune receptors that recognize pathogen effectors and trigger robust defense responses, a mechanism known as effector-triggered immunity (ETI) [64] [27]. The evolutionary significance of NLRs spans the diversity of land plants, from early-diverging lineages like mosses and ferns to modern angiosperms.
Mosses, representing some of the earliest terrestrial plants, provide invaluable insights into the evolution of plant immunity. These non-vascular plants lack traditional vascular systems yet demonstrate remarkable resistance to many pathogens that affect vascular plants [65]. Research on the model moss Physcomitrella patens has revealed that despite their simple morphology, mosses possess conserved NLR-mediated immune mechanisms shared with angiosperms [65]. Ferns, occupying an evolutionary position between bryophytes and seed plants, further bridge this gap, encoding diverse NLRs including sub-families lost in flowering plants [66].
This whitepaper examines how differential NLR responses contribute to disease resistance across plant species, focusing on comparative studies between resistant and susceptible cultivars. Understanding these mechanisms from an evolutionary perspective provides valuable genetic resources and strategies for enhancing disease resistance in crop plants.
Evolutionary Conservation of NLR-Mediated Immunity
NLR Immune Mechanisms Across Land Plants
The innate immune system of plants comprises two interconnected layers: pattern-triggered immunity (PTI) at the cell surface and effector-triggered immunity (ETI) intracellularly mediated by NLRs. While PTI provides broad-spectrum resistance against conserved microbial patterns, ETI offers specific recognition of pathogen effectors, often resulting in a hypersensitive response (HR) that limits pathogen spread [67]. This NLR-mediated immunity shows remarkable conservation across land plants despite 450 million years of evolution.
Studies in mosses have revealed that they exhibit resistance to many pathogens that typically affect vascular plants, employing distinct chemical and physical defense mechanisms while sharing fundamental innate immune systems common to all land plants [65]. Ferns, which diverged before seed plants, possess a diverse repertoire of NLRs including TIR-NLRs, CC-NLRs, and RPW8-NLRs, but not the bryophyte-specific Kin-NLRs and Hyd-NLRs found in mosses [66]. This pattern reflects the evolutionary trajectory of NLR genes, with certain lineages expanding while others were lost in specific plant groups.
The conservation of key molecular features in NLR proteins across evolutionary time underscores their fundamental role in plant immunity. Functionally conserved sequence motifs, such as the P-loop and MHD motifs within the central NB-ARC domain, are critical for NLR function [27]. Mutations in these regions can render NLRs nonfunctional or autoactive, demonstrating their essential role in immune signaling [27].
Methodological Framework for Comparative NLR Analysis
To systematically study NLR evolution and function across plant species, researchers employ a standardized computational pipeline for phylogenomic analysis. The following workflow illustrates the key steps in identifying conserved sequence motifs in NLR proteins across diverse plant species:
Figure 1: Workflow for identifying conserved NLR motifs across plant species.
This phylogenomic approach allows researchers to identify molecular signatures that have remained conserved in the NLR gene family over evolutionary time, providing insights into functionally important regions [27]. The pipeline can be applied to NLR datasets from diverse plant species, from mosses to dicots, enabling comparative analyses of NLR evolution and function.
Comparative Analysis of NLR Profiles in Resistant vs. Susceptible Cultivars
Sorghum Anthracnose Resistance Case Study
A compelling example of differential NLR responses comes from sorghum anthracnose, caused by the fungal pathogen Colletotrichum sublineola. Through systematic disease assays on 365 sorghum accessions, researchers identified BTx623 as a resistant cultivar and Guojiaohong1 (GJH1) as a susceptible cultivar [64]. Genomic analysis revealed substantial differences in their NLR repertoires, as summarized in the table below:
Table 1: Comparative NLR profiles in anthracnose-resistant and susceptible sorghum cultivars
| Feature | Resistant Cultivar (BTx623) | Susceptible Cultivar (GJH1) |
|---|---|---|
| Total NLR Genes | 302 | 239 |
| NLRs on Chromosome 5 | 98 (32.45%) | Not reported |
| Expression during Infection | Higher number of highly expressed and inducible NLR genes | Fewer highly expressed NLR genes |
| NLR Clusters | 213 NLR genes within 200 kb clusters | Not reported |
| Paired NLRs | 11 pairs identified | Not reported |
| Atypical Domains | 20 NLRs with integrated domains | Not reported |
The resistant cultivar BTx623 possesses 302 NLR genes, substantially more than the 239 identified in the susceptible GJH1 [64]. This disparity in NLR count suggests that a more diverse NLR repertoire may contribute to broader pathogen recognition capabilities. Furthermore, NLR genes were unevenly distributed across chromosomes, with chromosome 5 containing the highest number (98 NLRs, 32.45% of the total) in BTx623 [64].
During C. sublineola infection, BTx623 exhibited a higher number of highly expressed and inducible NLR genes compared to GJH1 [64]. This differential expression profile suggests that the resistant cultivar mounts a more robust transcriptional immune response, potentially enabling more effective pathogen recognition and defense activation.
Structural and Functional Diversity of NLR Proteins
Beyond quantitative differences, NLR proteins exhibit remarkable structural diversity that impacts their function. In the resistant sorghum cultivar BTx623, NLRs were classified into four categories based on their domain architecture:
Table 2: Classification of NLR proteins in the resistant sorghum cultivar BTx623
| NLR Category | Domain Architecture | Number in BTx623 | Proposed Function |
|---|---|---|---|
| CNL | CC-NBS-LRR | 187 | Full-length immune receptors |
| CN | CC-NBS | 62 | Signaling components or helpers |
| NL | NBS-LRR | 35 | Sensor NLRs |
| N | NBS only | 18 | Signaling intermediates |
Among the 302 NLRs in BTx623, 20 contained at least one atypical NLR domain or integrated domain (ID), representing 13 distinct Pfam domains including Pkinase_Tyr, WD40, FNIP, and WRKY [64]. These integrated domains potentially function as baits for pathogen effectors, expanding the recognition capabilities of the plant immune system.
NLR genes in BTx623 were predominantly located on chromosome arms and telomeric regions, with fewer in pericentromeric and centromeric regions [64]. Researchers identified 213 NLR genes located within 200 kb clusters, as well as 11 pairs of adjacent NLR genes that may work together to detect pathogen effectors and activate immune responses [64]. Such genomic organization facilitates the evolution of new recognition specificities through recombination and gene conversion.
Experimental Protocols for NLR Characterization
Genomic Identification and Annotation of NLR Genes
Comprehensive characterization of NLR immune receptors begins with systematic genome-wide identification and annotation. The following protocol outlines key steps for extracting NLR genes from plant proteomes:
- Data Acquisition: Download protein sequence files from reference genome databases. For comparative analysis, compile proteomes from multiple plant species into a single FASTA file.
- NLR Annotation: Annotate NLRs from the input protein sequence file using specialized tools such as NLRtracker or NLR-Annotator. NLRtracker, which utilizes InterProScan for protein function characterization, is recommended for its sensitivity and accuracy in extracting NLRs from plant proteomes [27].
- Sequence Curation: Manually curate the output to remove false positives and ensure comprehensive NLR identification using the NB-ARC Pfam model PF00931 as reference [64].
- Classification: Categorize NLRs into subfamilies (CNL, CN, NL, N) based on the presence of coiled-coil (CC), nucleotide-binding (NBS), and leucine-rich repeat (LRR) domains [64].
This protocol can be applied to genomic data from diverse plant species, enabling comparative analyses of NLR repertoires across the plant kingdom, from mosses to dicots.
Expression Profiling of NLR Genes During Pathogen Stress
Measuring NLR transcriptional dynamics in response to pathogen infection is crucial for understanding differential immune responses. The following workflow outlines the experimental process for expression profiling:
Figure 2: Workflow for NLR expression profiling under pathogen stress.
Key methodological considerations include:
- Plant Material Selection: Use genetically characterized resistant and susceptible cultivars, such as BTx623 and GJH1 for sorghum anthracnose or 'Nanane' and 'Misugi' for Brassica rapa white rust [64] [67].
- Pathogen Inoculation: Apply standardized inoculation methods appropriate for the pathosystem, such as spray inoculation for foliar pathogens or punch inoculation for localized infection [64].
- Time-Course Sampling: Collect tissue samples at multiple time points after inoculation (e.g., 0, 24, 48, 72 hours after inoculation) to capture dynamic expression changes [67].
- RNA Sequencing: Extract total RNA using validated systems (e.g., SV Total RNA Isolation System), prepare sequencing libraries, and perform high-throughput sequencing [67].
- Bioinformatic Analysis: Identify differentially expressed genes (DEGs) between inoculated and non-inoculated samples, with particular focus on NLR genes and defense-related pathways [67].
This approach revealed that in response to Albugo candida infection, resistant Brassica rapa cultivars upregulate salicylic acid-dependent systemic acquired resistance genes, while susceptible cultivars show different expression patterns [67].
Table 3: Essential research reagents and computational tools for NLR characterization
| Category/Reagent | Specific Tool/Resource | Application/Function | Key Features |
|---|---|---|---|
| NLR Annotation Tools | NLRtracker v1.0.3 | NLR identification from proteome data | High sensitivity/accuracy, uses InterProScan |
| NLR-Annotator v2.1 | NLR identification from nucleotide sequences | Works with nucleotide sequences | |
| Sequence Analysis | InterProScan 5.53-87.0 | Protein function characterization | Domain identification, functional prediction |
| MAFFT v7 | Multiple sequence alignment | Handles large datasets | |
| HMMER v3.4 | Sequence homology search | Profile HMM-based searches | |
| Phylogenetic Analysis | RAxML v8.2.12 | Maximum likelihood phylogenetic trees | Handles large phylogenetic trees |
| iTOL | Tree visualization and annotation | Interactive tree visualization | |
| Motif Discovery | MEME Suite v5.5.5 | Motif-based sequence analysis | De novo motif discovery |
| Clustering Algorithms | MCL v14-137 | Protein sequence clustering | Markov clustering algorithm |
| Experimental Validation | Nicotiana benthamiana | Heterologous system for NLR function testing | Hypersensitive response assay |
This toolkit enables researchers to comprehensively characterize NLR immune receptors from genomic identification to functional validation. The integration of computational and experimental approaches provides a powerful framework for elucidating NLR function in plant immunity.
The comparative analysis of NLR immune receptors in resistant versus susceptible plant cultivars reveals a complex landscape of genomic, transcriptional, and structural differences that underlie disease outcomes. Resistant cultivars typically possess more diverse NLR repertoires, exhibit stronger induction of NLR genes upon pathogen challenge, and maintain specific NLR architectures that may enhance pathogen recognition capabilities.
From an evolutionary perspective, the conservation of NLR-mediated immunity from mosses to dicots underscores the fundamental importance of this defense mechanism across 450 million years of plant evolution. While core NLR structure and function are maintained, lineage-specific expansions and innovations have shaped the immune repertoire of different plant groups, resulting in distinct disease resistance strategies.
Future research directions should include comprehensive comparative analyses of NLR evolution across broader plant lineages, functional characterization of non-canonical NLRs with integrated domains, and development of precision breeding strategies that leverage natural NLR diversity. The integration of phylogenomics, structural biology, and genome editing will further advance our ability to harness NLR genes for crop improvement, ultimately contributing to global food security.
Nucleotide-binding leucine-rich repeat (NLR) proteins constitute the largest and most variable family of plant disease-resistance (R) genes, serving as critical intracellular immune receptors that mediate effector-triggered immunity (ETI) [68] [37]. These proteins exhibit a characteristic tripartite domain architecture: a central nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4 (NB-ARC) domain, a C-terminal leucine-rich repeat (LRR) domain, and variable N-terminal domains that classify NLRs into major subclasses including TIR-NBS-LRR (TNL), CC-NBS-LRR (CNL), and RPW8-NBS-LRR (RNL) [1] [69]. The N-terminal domains typically function as signaling modules, the NB-ARC domain acts as a molecular switch regulated by ADP/ATP exchange, and the LRR domain is primarily involved in pathogen recognition and autoinhibition [69] [37].
The evolution of NLR genes across land plants, from mosses to dicots, reveals a dynamic history of expansion, contraction, and functional specialization. NLRs originated in the common ancestor of all green plants, with identified homologs in green algae and bryophytes [70]. In flowering plants, NLRs have undergone significant lineage-specific expansions, resulting in substantial variation in gene numbers between species—from approximately 50 in papaya to over 1,000 in apple and hexaploid wheat [37]. This expansion has been driven primarily by tandem gene duplication events, leading to the formation of NLR clusters in specific genomic regions [71] [42]. Throughout plant evolution, NLRs have evolved from single individual genetic units ("singletons") to higher-order configurations, including sensor-helper NLR pairs and complex networks, enabling more robust and adaptable immune responses [37].
Protein interaction networks are fundamental to NLR function, with many NLRs operating in interconnected pairs or networks where sensor NLRs detect pathogen effectors and helper NLRs transduce immune signals [37]. Understanding these networks, identifying key hub proteins within them, and mapping the signaling pathways they activate represents a critical frontier in plant immunity research, with implications for engineering disease resistance in crops.
Evolutionary Context of NLR Networks in Land Plants
Evolutionary Trajectory from Early Plants to Dicots
The NLR gene family demonstrates remarkable evolutionary dynamism across plant lineages. In basal land plants such as bryophytes (e.g., the moss Physcomitrella patens), NLR repertoires are relatively small, with approximately 25 members identified [1]. These early NLRs already exhibited domain diversification, including the presence of TNL, CNL, and unique subclasses such as HNL (α/β-hydrolase-NBS-LRR) in liverworts and PNL (protein-kinase-NBS-LRR) in mosses [70]. The transition to vascular plants was marked by a significant expansion of NLR genes, with lycophytes like Selaginella moellendorffii showing a surprisingly minimal NLR repertoire of only 2 genes, suggesting lineage-specific contraction [1].
In angiosperms, NLR evolution proceeded in two distinct stages: an initial period of relatively stable, low gene numbers from the origin of angiosperms until the Cretaceous-Paleogene (K-Pg) boundary, followed by a dramatic expansion that led to the extensive NLR diversity observed in contemporary species [70]. This expansion was not uniform across lineages, with different plant families exhibiting distinct evolutionary patterns—Brassicaceae show "first expansion and then contraction," Poaceae display "contraction," while Fabaceae and Rosaceae demonstrate consistently expanding patterns [70]. Magnoliids, occupying a critical phylogenetic position as early-diverging angiosperms, provide important insights into NLR evolution, with some species showing complete absence of TNL genes, presumably due to immune pathway deficiencies [70].
Molecular Mechanisms Driving NLR Network Evolution
The evolution of NLR networks has been shaped by several molecular mechanisms that generate diversity and functional specialization:
Domain Rearrangement and Integration: NLR evolution has involved extensive domain shuffling, with acquisition of novel integrated domains (IDs) that often function as "decoys" for pathogen effectors [71]. These integrated domains, which can include WRKY, kinase, heavy metal-associated (HMA), and zinc-finger BED domains, enable direct recognition of pathogen effectors and expand the pathogen recognition capacity of the NLR network [68].
Tandem Duplication and Birth-and-Death Evolution: Tandem duplication serves as the primary mechanism for NLR expansion across angiosperms, leading to the formation of gene clusters that facilitate rapid generation of new resistance specificities [71] [42]. This process follows a birth-and-death evolutionary model, where new NLR genes are created through duplication and subsequently undergo divergent evolution or pseudogenization [71].
Helper-Sensor Specialization: A key innovation in NLR network evolution is the functional specialization into sensor NLRs (responsible for pathogen recognition) and helper NLRs (responsible for signal transduction) [37]. This division of labor enables the construction of complex immune networks where multiple sensor NLRs can connect to common helper NLRs, increasing both robustness and evolvability of the immune system [37].
Table 1: Evolutionary Patterns of NLR Genes Across Plant Lineages
| Plant Group | Representative Species | NLR Count | Dominant Subclasses | Key Evolutionary Features |
|---|---|---|---|---|
| Green Algae | Chlamydomonas reinhardtii | 0 | None | Absence of canonical NLRs [1] |
| Bryophytes | Physcomitrella patens | ~25 | TNL, CNL, HNL, PNL | Presence of lineage-specific subclasses [70] |
| Lycophytes | Selaginella moellendorffii | ~2 | Limited diversity | Extreme NLR contraction [1] |
| Monocots | Oryza sativa (rice) | ~498 | CNL, RNL | Absence of TNLs [70] |
| Basal Eudicots | Aquilegia coerulea | Variable | CNL, TNL, RNL | Lineage-specific expansions [21] |
| Magnoliids | Litsea cubeba, Persea americana | 70-400+ | Primarily CNL | Multiple independent TNL losses [70] |
| Eudicots | Arabidopsis thaliana | 165 | TNL, CNL, RNL | Balanced subclass representation [70] |
Predicting NLR Interaction Networks and Hub Proteins
Genomic and Computational Approaches
Predicting NLR interaction networks begins with comprehensive identification and annotation of NLR repertoires in plant genomes. The standard workflow involves:
Genome-Wide NLR Identification: Using hidden Markov model (HMM)-based searches with NB-ARC domain profiles (PF00931) against plant proteomes, followed by validation with tools like NLR-Annotator [72]. Additional BLASTp searches with known NLR proteins from model species (e.g., Arabidopsis thaliana) can supplement this approach [42].
Domain Architecture Annotation: Classifying identified NLRs into subclasses (TNL, CNL, RNL) based on N-terminal domains using tools like CD-Search and InterProScan, while also identifying integrated domains that may function in effector recognition [68] [42].
Phylogenetic Analysis: Constructing maximum likelihood phylogenetic trees using NB-ARC domains or full-length protein sequences to elucidate evolutionary relationships and identify conserved clades [72] [42].
Genomic Distribution Mapping: Analyzing chromosomal locations to identify NLR-rich regions and clusters, which often represent hotspots for immune gene evolution [71] [42].
Network Prediction Methodologies
Several computational approaches enable prediction of NLR protein interaction networks:
Co-Expression Network Analysis: Using transcriptome data from pathogen infections or immune elicitation to identify NLR genes with correlated expression patterns, suggesting functional relationships [73] [42]. Weighted Gene Co-expression Network Analysis (WGCNA) is particularly useful for this purpose.
Protein-Protein Interaction Prediction: Utilizing tools like STRING-db that integrate multiple evidence sources including genomic context, gene fusion events, co-expression, and literature mining to predict potential interactions [42].
Evolutionary Rate Correlation: Analyzing correlated evolutionary rates between NLR genes, as proteins that interact often show correlated evolutionary signatures due to co-evolution.
Promoter cis-Element Analysis: Identifying shared regulatory elements in NLR promoters that suggest co-regulation and potential functional relationships [42].
Table 2: Experimentally Validated NLR Hub Proteins in Plant Immunity
| Hub Protein | Species | NLR Class | Network Role | Validated Interactions | Key References |
|---|---|---|---|---|---|
| Caz01g22900 | Capsicum annuum (pepper) | CNL | Putative signaling hub | Interacts with multiple NLRs; highly expressed during Phytophthora infection [42] | [42] |
| Caz09g03820 | Capsicum annuum (pepper) | CNL | Putative hub in PPI network | Central node in protein interaction network; responsive to pathogen infection [42] | [42] |
| NRC family | Solanaceae species | CNL | Helper NLR hub | Functions as common helper for multiple sensor NLRs [68] | [68] |
| ADR1 | Arabidopsis thaliana | RNL | Helper NLR | Required for TNL signaling; forms network with multiple TNL sensors [37] | [37] |
| ZAR1 | Arabidopsis thaliana | CNL | Resistosome core | Oligomerizes to form pentameric hub for immune signaling [68] | [68] |
Diagram 1: NLR Protein Interaction Network Architecture. This diagram illustrates the complex network relationships between sensor NLRs, helper NLR hubs, pathogen effectors, and guardee/decoy proteins in plant immunity.
Experimental Validation of NLR Interactions and Pathways
Protein-Protein Interaction Assays
Several well-established experimental approaches enable validation of predicted NLR interactions:
Yeast Two-Hybrid (Y2H) Systems remain a cornerstone for binary protein interaction validation. For NLR studies, specialized Y2H approaches are often required due to the auto-activation potential of NLR domains. Protocol: (1) Clone full-length or truncated NLR genes into both bait (DNA-binding domain) and prey (activation domain) vectors; (2) Transform into appropriate yeast strains (e.g., AH109); (3) Plate on selective media (-Leu/-Trp/-His/-Ade) with X-α-Gal to test for interactions; (4) Quantify interactions using β-galactosidase assays [69].
Co-Immunoprecipitation (Co-IP) followed by Mass Spectrometry provides evidence for in vivo interactions and identifies complex components. Protocol: (1) Express epitope-tagged NLR proteins in plant systems (e.g., Nicotiana benthamiana); (2) Extract proteins under native conditions; (3) Immunoprecipitate using tag-specific antibodies; (4) Analyze co-precipitating proteins by Western blot or identify unknown interactors by LC-MS/MS [69].
Bimolecular Fluorescence Complementation (BiFC) visualizes protein interactions in living plant cells. Protocol: (1) Fuse NLR genes to split YFP fragments (YN/YF); (2) Co-express in plant cells via transient transformation; (3) Visualize fluorescence complementation by confocal microscopy 24-48 hours post-infiltration [69].
Functional Validation of NLR Hubs
Genetic Approaches are essential for establishing the functional significance of NLR hubs:
Knockout/Knockdown Studies: Using CRISPR-Cas9 or RNAi to disrupt putative hub NLRs and evaluating the impact on overall immune signaling. For example, knockout of helper NLR hubs like NRC proteins in solanaceous plants abolishes immunity mediated by multiple sensor NLRs [68].
Heterologous Expression: Expressing candidate hub NLRs in susceptible species or non-host plants to test for sufficiency in conferring resistance. This approach demonstrated that some NLR hubs can function across plant families, indicating evolutionary conservation of signaling mechanisms [1].
Biochemical Assays probe the mechanistic basis of hub function:
ATPase Activity Assays: Measuring nucleotide binding and hydrolysis activities of NLR NB-ARC domains, as hub activation typically involves ADP/ATP exchange [37].
Oligomerization Studies: Using size-exclusion chromatography, crosslinking, and native gels to detect hub-dependent oligomerization events, such as the formation of resistosomes [68] [37].
Structural Studies: X-ray crystallography and cryo-EM of NLR hubs have revealed fundamental mechanisms of action, such as the ZAR1 resistosome structure that forms a calcium-permeable channel upon activation [68].
Diagram 2: NLR Hub Validation Workflow. This diagram outlines the sequential experimental approaches for validating predicted NLR hub proteins and their interactions.
NLR Signaling Pathways and Network Dynamics
Core Signaling Mechanisms
NLR immune signaling involves sophisticated molecular mechanisms that convert pathogen perception into defense activation:
Oligomerization-Based Signaling: Upon effector recognition, many NLRs undergo nucleotide-dependent oligomerization to form signaling-competent complexes called resistosomes [68] [37]. CNL-type resistosomes (e.g., ZAR1) form calcium-permeable channels that initiate downstream signaling, while TNL-type resistosomes often exhibit enzymatic activities that produce small signaling molecules [69] [68].
Helper NLR Activation: Sensor NLRs typically activate helper NLRs through direct interaction or through intermediate signaling components. For TNLs, this often involves the EDS1 (Enhanced Disease Susceptibility 1) family proteins, which transduce signals to helper RNLs [69] [37]. CNLs can directly or indirectly activate helper NLRs from the NRC family in solanaceous plants or RNLs in Arabidopsis [68].
Transcriptional Reprogramming: Activated NLR networks initiate massive transcriptional reprogramming through both direct and indirect mechanisms. Some NLRs have been shown to interact with transcription factors or directly localize to the nucleus to regulate defense gene expression [73].
Network Properties and Dynamics
NLR immune networks exhibit several key properties that enhance their effectiveness:
Robustness: The use of helper NLR hubs that serve multiple sensor NLRs creates a robust system where loss of individual sensor components does not completely compromise immunity [37].
Signaling Amplification: Helper NLR hubs can amplify signals from weakly activated sensor NLRs, ensuring effective immune activation even when pathogen effectors are present at low concentrations [73].
Conditional Activation: NLR networks incorporate multiple regulatory mechanisms to prevent inappropriate activation in the absence of pathogens, including autoinhibitory interactions, chaperone-mediated regulation, and transcriptional control [73] [37].
Spatiotemporal Dynamics: NLR networks exhibit precise spatial and temporal regulation, with some components showing tissue-specific expression or rapid transcriptional induction upon infection [73] [70].
Table 3: Key Signaling Pathways in NLR Network Function
| Signaling Pathway | Key Components | NLR Classes Involved | Signaling Output | Evolutionary Conservation |
|---|---|---|---|---|
| TNL-EDS1-RNL Pathway | TNL sensors, EDS1, PAD4, SAG101, RNL helpers (NRG1/ADR1) | TNL, RNL | Transcriptional reprogramming, HR | Conserved in dicots [69] [37] |
| CNL Resistosome Channel | CNL sensors (e.g., ZAR1), helper NLRs | CNL | Calcium influx, MAPK activation, HR | Widely conserved [68] |
| NRC Network | Sensor CNLs, NRC helpers | CNL | HR, defense gene expression | Solanaceae-specific [68] |
| Integrated Domain Signaling | NLR-IDs, effector targets | TNL, CNL | Direct effector recognition, HR | Lineage-specific [68] [37] |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for NLR Network Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Bioinformatics Tools | NLR-Annotator, HMMER, OrthoFinder2, MCScanX | Genome-wide NLR identification, classification, evolutionary analysis | Requires genomic data; validation needed [72] [42] |
| Interaction Validation Systems | Yeast Two-Hybrid, Co-IP kits, BiFC vectors | Protein-protein interaction detection | Watch for auto-activation with NLRs [69] [42] |
| Heterologous Expression Systems | Nicotiana benthamiana, protoplasts, Arabidopsis mutants | Functional testing of NLR genes | Consider species-specific requirements [69] [68] |
| Genetic Manipulation Tools | CRISPR-Cas9, RNAi vectors, T-DNA mutants | Loss-of-function studies | Redundancy may mask phenotypes [73] [42] |
| Pathogen Assays | Pseudomonas syringae, Hyaloperonospora, Phytophthora | Functional immune response analysis | Match appropriate pathogens to NLRs [69] [42] |
| Biochemical Assay Kits | ATPase activity, crosslinkers, membrane potential dyes | Mechanistic studies of NLR function | Optimize for plant-specific conditions [68] [37] |
| Transcriptional Reporters | Defense gene promoters, GUS, luciferase | Monitoring immune activation | Multiple reporters recommended [73] [42] |
Future Directions and Technical Challenges
The study of NLR protein interaction networks faces several technical challenges that represent opportunities for methodological advancement. The extensive genetic redundancy within NLR networks often masks phenotypic effects in loss-of-function studies, necessitating development of higher-order mutants and conditional knockout systems [37]. The transient and conditional nature of many NLR interactions requires improved methods for capturing dynamic protein complexes in living plant cells. The size and repetitive nature of NLR genes presents challenges for genetic manipulation, calling for optimized transformation and gene editing protocols [42].
Emerging technologies are poised to overcome these limitations. Single-cell transcriptomics will reveal cell-type-specific expression patterns of NLR network components [73]. Advanced microscopy techniques, including FRET-FLIM and super-resolution imaging, will enable visualization of NLR complex formation and dynamics in real time [69]. Structural biology approaches, particularly cryo-electron tomography, may capture NLR resistosomes in their native membrane environment [68] [37]. Proteomics methods such as proximity labeling (e.g., TurboID) can map NLR interactions under resting and activated states [37].
From an evolutionary perspective, integrating comparative genomics across diverse plant species with functional studies will reveal how NLR networks have been rewired throughout plant evolution and identify core conserved principles versus lineage-specific innovations [21] [70]. Such insights will guide engineering of synthetic NLR networks with novel recognition specificities and enhanced signaling properties for crop improvement.
Nucleotide-binding leucine-rich repeat (NLR) genes encode intracellular immune receptors that constitute a cornerstone of the plant innate immune system, mediating effector-triggered immunity (ETI) upon pathogen recognition [37]. These genes represent one of the most diverse and rapidly evolving gene families in plants, reflecting the continuous evolutionary arms race between plants and their pathogens [74] [37]. The functional validation of NLR genes is therefore critical for understanding plant immunity and for engineering disease-resistant crops. This technical guide focuses on two pivotal methodologies—Virus-Induced Gene Silencing (VIGS) and Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)—that enable researchers to confirm NLR activity and elucidate their functional evolution across land plants, from early diverging mosses to advanced dicots [74].
The Molecular Basis of NLR Function
NLR Domain Architecture and Classification
NLR proteins exhibit a characteristic tripartite domain architecture, functioning as molecular switches within the plant cell [37]. The central nucleotide-binding domain (NB-ARC or NBS) facilitates conformational changes through ADP/ATP exchange, while the C-terminal leucine-rich repeat (LRR) domain is often involved in effector recognition and autoinhibition. The N-terminal domain, which can be a coiled-coil (CC), toll/interleukin-1 receptor (TIR), or RPW8-like domain, dictates downstream signaling pathways [37]. This modular structure allows NLRs to detect pathogen effectors directly or indirectly through integrated decoy domains, leading to immune activation often accompanied by a hypersensitive response [62] [37].
Evolutionary Dynamics of NLR Genes
The NLR gene family has undergone remarkable expansion and diversification throughout plant evolution. Comparative genomics reveals that NLRs are present in all major plant lineages, from green algae to angiosperms, with significant variation in repertoire size and complexity [74] [37]. While bryophytes like Physcomitrella patens possess relatively small NLR repertoires (approximately 25 NLRs), flowering plants often harbor hundreds to over a thousand NLR genes, driven primarily by tandem duplication and whole-genome duplication events [74] [75] [37]. This expansion enables plants to recognize the rapidly evolving effector repertoires of diverse pathogens, with lineage-specific expansions reflecting adaptations to particular pathogenic threats [76] [75].
Experimental Workflows for NLR Functional Validation
The following diagrams illustrate the integrated experimental pipelines for validating NLR gene function using VIGS and RT-qPCR, contextualized within evolutionary studies.
VIGS Workflow for NLR Functional Analysis
Diagram 1: VIGS workflow for NLR function validation. This process enables rapid assessment of NLR gene function by specifically silencing candidate genes and evaluating the resulting changes in disease resistance phenotypes.
RT-qPCR Workflow for NLR Expression Analysis
Diagram 2: RT-qPCR workflow for NLR expression analysis. This methodology provides quantitative assessment of NLR gene expression patterns in response to pathogen challenge or across different plant lineages.
Core Techniques: Principles and Applications
Virus-Induced Gene Silencing (VIGS)
VIGS is a powerful reverse genetics tool that leverages the plant's natural RNA-mediated antiviral defense system to transiently silence target genes. When applied to NLR validation, VIGS allows researchers to directly link specific NLR genes to disease resistance phenotypes by knocking down their expression and observing the resulting susceptibility [74] [77] [78].
In practice, a 150-300 base pair fragment of the target NLR gene is cloned into a modified viral vector (such as Tobacco Rattle Virus or Barley Stripe Mosaic Virus). The recombinant vector is then introduced into plants via Agrobacterium-mediated infiltration or mechanical inoculation. As the virus spreads systemically, it triggers sequence-specific degradation of endogenous mRNA transcripts, leading to reduced expression of the target NLR gene [74] [78]. The effectiveness of this approach was demonstrated in cotton, where silencing of GaNBS (OG2) led to increased susceptibility to cotton leaf curl disease, confirming its essential role in virus resistance [74]. Similarly, VIGS validation of the wheat NLR gene TaRPM1-2D established its function in powdery mildew resistance [77].
Reverse Transcription Quantitative PCR (RT-qPCR)
RT-qPCR provides precise quantification of NLR transcript abundance, enabling researchers to correlate gene expression patterns with resistance phenotypes and understand NLR regulation in different evolutionary contexts. This technique involves converting mRNA to complementary DNA (cDNA) followed by quantitative PCR amplification with NLR-specific primers [75].
Key considerations for NLR expression analysis include proper normalization using validated reference genes (e.g., Actin, EF1α, UBQ) and rigorous primer design to account for the high sequence diversity among NLR family members. Differential expression analysis of NLR genes between resistant and susceptible genotypes, or before and after pathogen challenge, can identify candidates for functional validation [77] [75]. In pepper, RT-qPCR analysis identified 44 NLR genes significantly differentially expressed following Phytophthora capsici infection, highlighting potential key players in disease resistance [75].
Integrated Validation: Case Studies
Validating a Novel NLR in Wheat Powdery Mildew Resistance
The identification and validation of TaRPM1-2D in wheat cultivar 'Brock' exemplifies the powerful integration of these methodologies. Genetic mapping first localized the resistance to a 6.88 Mb interval on chromosome 2D, containing the highly expressed TaRPM1-2D gene encoding a typical NLR protein [77]. Expression analysis via RT-qPCR revealed significantly higher transcript levels of TaRPM1-2D in resistant 'Brock' and its near-isogenic line 'BJ-1' compared to susceptible 'Jing411'. Functional validation through VIGS-mediated silencing demonstrated that knocking down TaRPM1-2D expression compromised resistance, confirming its essential role in powdery mildew defense [77].
Cross-Species NLR Function in Wheat Rust Resistance
The transfer of the Yr87/Lr85 NLR gene from Aegilops sharonensis and Aegilops longissima to wheat demonstrates the practical application of these validation techniques across species boundaries. This unique NLR confers resistance against both stripe rust and leaf rust pathogens, an unusual breadth of specificity [78]. VIGS experiments targeting Yr87/Lr85 in resistant introgression lines reduced gene expression by >75% and rendered plants susceptible to both rust pathogens, confirming its necessity for resistance. This study highlights how NLR validation enables the exploitation of evolutionary diversity for crop improvement [78].
Research Reagent Solutions
Table 1: Essential research reagents for NLR functional validation
| Reagent/Category | Specific Examples | Function/Application in NLR Research |
|---|---|---|
| VIGS Vectors | Tobacco Rattle Virus (TRV), Barley Stripe Mosaic Virus (BSMV) | Delivery of NLR-specific sequences for targeted gene silencing [74] [78] |
| Reverse Transcriptases | M-MLV, AMV | cDNA synthesis from RNA templates for expression analysis [75] |
| qPCR Master Mixes | SYBR Green, TaqMan | Fluorescent detection of NLR amplification products [75] |
| Reference Genes | Actin, Ubiquitin, EF1α | Normalization of NLR expression data [75] |
| Cloning Systems | Gateway, Golden Gate | Assembly of NLR constructs for VIGS or overexpression [74] |
Quantitative Data from NLR Functional Studies
Table 2: Representative quantitative data from NLR validation studies
| NLR Gene | Plant Species | Validation Method | Key Quantitative Results | Pathogen System |
|---|---|---|---|---|
| GaNBS (OG2) | Gossypium arboreum (cotton) | VIGS | Silencing led to increased virus accumulation; putative role in virus tittering [74] | Cotton leaf curl disease [74] |
| TaRPM1-2D | Triticum aestivum (wheat) | VIGS, RT-qPCR | Expression significantly higher in resistant lines; silencing compromised resistance [77] | Powdery mildew (Blumeria graminis) [77] |
| Yr87/Lr85 | Aegilops sharonensis | VIGS, Mutational analysis | >75% reduction in expression via VIGS resulted in susceptibility [78] | Leaf rust (Puccinia triticina) & stripe rust (P. striiformis) [78] |
| 44 NLRs | Capsicum annuum (pepper) | RNA-seq, RT-qPCR | 44 NLRs significantly differentially expressed post-Phytophthora infection [75] | Phytophthora capsici [75] |
Technical Considerations and Best Practices
Experimental Design and Optimization
Successful NLR validation requires careful experimental design. For VIGS studies, target fragment selection is critical—regions with low similarity to other NLRs minimize off-target effects. Including multiple non-overlapping fragments for the same gene strengthens phenotypic correlations [78]. Optimal sampling timepoints post-inoculation (e.g., 0, 24, 48 hours) capture dynamic NLR expression patterns during early defense responses [78]. Experimental controls must include empty vector controls, untreated plants, and appropriate reference genes validated for the specific plant system [75].
Challenges and Solutions in NLR Studies
The high diversity and sequence similarity among NLR genes present technical challenges. Paralogous genes may share significant homology, complicating specific silencing or quantification. This can be addressed through careful primer/probe design targeting unique regions and validating specificity via sequencing of amplification products. The frequent clustering of NLR genes in plant genomes can lead to co-silencing of multiple NLRs in VIGS experiments, potentially confounding results. Solutions include using shorter gene-specific fragments and verifying silencing specificity through sequencing of VIGS products [74] [78].
VIGS and RT-qPCR represent indispensable tools in the functional validation of NLR genes, providing complementary approaches to establish gene-phenotype relationships and elucidate expression dynamics. When applied within an evolutionary framework, these techniques illuminate how NLR diversity has been generated, maintained, and selected throughout plant evolution. The continued refinement of these methodologies, coupled with emerging technologies in genomics and genome editing, will accelerate the discovery and deployment of NLR genes for crop improvement, ultimately enhancing agricultural sustainability in the face of evolving pathogen threats.
Conclusion
The evolutionary journey of NLR genes, from the compact repertoire of mosses to the vast, complex families in flowering plants, underscores a continuous molecular arms race with pathogens. Key takeaways reveal that NLR diversification is driven by dynamic processes like tandem duplication and is finely regulated to balance robust immunity with plant fitness. Modern methodologies, from pangenomics to high-throughput functional screening, are rapidly accelerating the discovery of novel NLRs. However, comparative genomics also warns of NLR erosion during domestication, highlighting the critical need to preserve wild genetic resources. The future of plant health and sustainable agriculture lies in leveraging these evolutionary insights. Translational applications include engineering NLRs with expanded recognition spectra and deploying optimized NLR stacks to provide resilient, broad-spectrum disease resistance, a principle with profound implications for securing global food systems.
References
- 1. Evolution and Conservation of Plant NLR Functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3782705/]
- 2. Plant NLR diversity: the known unknowns of pan-NLRomes [https://pmc.ncbi.nlm.nih.gov/articles/PMC8226294/]
- 3. The evolution of plant NLR immune receptors and ... [https://www.sciencedirect.com/science/article/abs/pii/S1369526623000286]
- 4. Structural basis of NLR activation and innate immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8813719/]
- 5. Comparative analysis of the NLR gene family in the genomes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507765/]
- 6. Diversity, abundance, and domain architecture of plant ... [https://www.sciencedirect.com/science/article/pii/S2405844024105063]
- 7. Comparative-genomic analysis reveals dynamic NLR gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10017999/]
- 8. Plant NLR Origins Traced Back to Green Algae [https://pubmed.ncbi.nlm.nih.gov/29887275/]
- 9. TIR-NBS-LRR genes are rare in monocots - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC2763876/]
- 10. DaapNLRSeek, prediction, and evolution of resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12226392/]
- 11. An NLR pair in the Pm68 locus confers powdery mildew ... [https://www.nature.com/articles/s41467-025-64048-z]
- 12. The Physcomitrella patens chromosome-scale assembly ... [https://pubmed.ncbi.nlm.nih.gov/29237241/]
- 13. The large repertoire of conifer NLR resistance genes ... [https://www.nature.com/articles/s41598-019-47950-7]
- 14. Comparative analysis of plant immune receptor architectures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4759884/]
- 15. Comparative analysis of plant immune receptor architectures ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-016-0228-7]
- 16. Tracing the path from conservation to expansion evolutionary ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-06233-2]
- 17. Physcomitrium patens - Genome assembly [https://plants.ensembl.org/Physcomitrium_patens/Info/Index]
- 18. Pangenomic context reveals the extent of intraspecific plant ... [https://www.sciencedirect.com/science/article/pii/S1931312825002835]
- 19. Comparative analysis, diversification, and functional ... [https://www.nature.com/articles/s41598-024-62876-5]
- 20. Genome-wide identification of the NLR gene family in ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08334-w]
- 21. An angiosperm NLR Atlas reveals that NLR gene reduction ... [https://www.sciencedirect.com/science/article/pii/S1674205221003142]
- 22. Vitis labrusca genome assembly reveals diversification ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2023.1234130/full]
- 23. Comparative Genomics and Functional Studies of Wheat ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7761493/]
- 24. Pangenomic context reveals the extent of intraspecific plant ... [https://pubmed.ncbi.nlm.nih.gov/40812179/]
- 25. Rapid evolution in a plant-pathogen interaction and the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3352484/]
- 26. Divergent evolution of NLR genes in the genus Glycine [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2024.1383135/full]
- 27. Phylogenomics of Plant NLR Immune Receptors to Identify ... [https://en.bio-protocol.org/en/bpdetail?id=5023&type=0]
- 28. NLR-parser: rapid annotation of plant NLR complements [https://pmc.ncbi.nlm.nih.gov/articles/PMC4426836/]
- 29. Genome-wide identification and gene expression analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12361222/]
- 30. Regulation and Evolution of NLR Genes - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC6032283/]
- 31. Variation Patterns of NLR Clusters in Arabidopsis thaliana ... [https://www.sciencedirect.com/science/article/pii/S2590346220301127]
- 32. High intraspecies allelic diversity in Arabidopsis NLR immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9882162/]
- 33. Moss-pathogen interactions: a review of the current status ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1539311/full]
- 34. An approach for prioritizing candidate genes from RNA-seq ... [https://pubmed.ncbi.nlm.nih.gov/37433118/]
- 35. Discovery of functional NLRs using expression level, high-throughput transformation and large-scale phenotyping | Nature Plants [https://www.nature.com/articles/s41477-025-02110-w]
- 36. Unlocking Functional NLRs via Expression and Phenotyping [https://bioengineer.org/unlocking-functional-nlrs-via-expression-and-phenotyping/]
- 37. NLR receptors in plant immunity: making sense of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10561179/]
- 38. NLRSeek: A reannotation–based pipeline for mining ... [https://www.sciencedirect.com/science/article/pii/S2662173825002085]
- 39. Engineering of autoactive NLRs: a big step toward ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12367612/]
- 40. Hands-on: Functional of protein annotation / sequences ... Functional [https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/functional/tutorial.html]
- 41. Regulation and Evolution of NLR Genes [https://www.mdpi.com/1422-0067/19/6/1662]
- 42. Genome-Wide Identification and Functional Evolution of ... [https://www.mdpi.com/1467-3045/47/10/867]
- 43. Oligomerization-mediated autoinhibition and cofactor ... [https://www.nature.com/articles/s41586-024-07668-7]
- 44. Evolution and Conservation of Plant NLR Functions [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2013.00297/full]
- 45. Transcript-level expression control of plant NLR genes [https://pubmed.ncbi.nlm.nih.gov/28834153/]
- 46. Epigenetically poised chromatin states regulate PRR and ... [https://link.springer.com/article/10.1007/s42994-025-00233-4]
- 47. Functional diversification gave rise to allelic specialization ... [https://elifesciences.org/articles/71662]
- 48. Phylogenomics of Plant NLR Immune Receptors to Identify ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11237980/]
- 49. Deciphering Plant NLR Genomic Evolution: Synteny-Informed Classification Unveils Insights into TNL Gene Loss - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11789945/]
- 50. Plant NLR receptor proteins and their potential in the ... [https://www.sciencedirect.com/science/article/pii/S2452072119301443]
- 51. Harnessing effector-triggered immunity for durable disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5810938/]
- 52. Molecular mechanisms that limit the costs of NLR‐mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6638094/]
- 53. Homosporous Land Plants [https://www.mobot.org/mobot/research/apweb/orders/sporing.html]
- 54. Domestication Reduces Plant Immune Receptor Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12342797/]
- 55. Evolution of NLR genes in genus Arachis reveals ... [https://www.nature.com/articles/s41598-023-36302-1]
- 56. Macroevolution of NLR genes in family Fabaceae provides ... [https://www.sciencedirect.com/science/article/pii/S2667064X23001215]
- 57. Conservation of NLR-triggered immunity across plant lineages [https://pmc.ncbi.nlm.nih.gov/articles/PMC3523848/]
- 58. Comparative analysis of the NLR gene family in ... [https://ui.adsabs.harvard.edu/abs/2025FrPS...1681919S/abstract]
- 59. Comparative analysis of the NLR gene family in ... [https://www.frontiersin.org/articles/10.3389/fpls.2025.1681919]
- 60. Chromosome-scale haplotype-phased genome assemblies of ... [https://www.semanticscholar.org/paper/Chromosome-scale-haplotype-phased-genome-assemblies-Shirasawa-Ueta/f0c9bdff1f7960e211f1aaf27c9b886fb19f3db6]
- 61. Investigation of NLR Genes Reveals Divergent Evolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10138078/]
- 62. NLRscape: an atlas of plant NLR proteins - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9825502/]
- 63. Comparative analysis of the NLR gene family in ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1681919/full]
- 64. Comparative genomic analysis reveals the difference of NLR immune... [https://phytopatholres.biomedcentral.com/articles/10.1186/s42483-025-00318-4]
- 65. Moss-pathogen interactions: a review of the current status ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11850516/]
- 66. Exploring fern pathosystems and immune receptors to bridge ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02413-6]
- 67. in the transcriptional immune Differences to Albugo candida... response [https://www.nature.com/articles/s41598-023-35205-5?error=cookies_not_supported&code=9a77fc3f-9ba0-4e05-bfab-8ff0b615d484]
- 68. Plant NLRs: Evolving with pathogen effectors and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9554439/]
- 69. Direct recognition of pathogen effectors by plant NLR immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9528080/]
- 70. Evolution of NLR Resistance Genes in Magnoliids [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.777157/full]
- 71. Multilevel evolution shapes the function of NB-LRR ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.1007288/full]
- 72. Diversity and Evolution of NLR Genes in Citrus Species [https://pmc.ncbi.nlm.nih.gov/articles/PMC11504038/]
- 73. The Ups and Downs of Plant NLR Expression During ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.921148/full]
- 74. Comparative analysis, diversification, and functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11126693/]
- 75. Genome-Wide Identification and Functional Evolution of NLR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562843/]
- 76. Tracing the path from conservation to expansion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11863892/]
- 77. Genetic Basis Identification of a NLR Gene, TaRPM1-2D, ... [https://www.mdpi.com/2223-7747/14/17/2652]
- 78. A single NLR gene confers resistance to leaf and stripe rust ... [https://www.nature.com/articles/s41467-024-54068-6]