A Practical Guide to Visual CRISPR Screening: Using GFP Reporters for Efficient Transformat Selection and Analysis
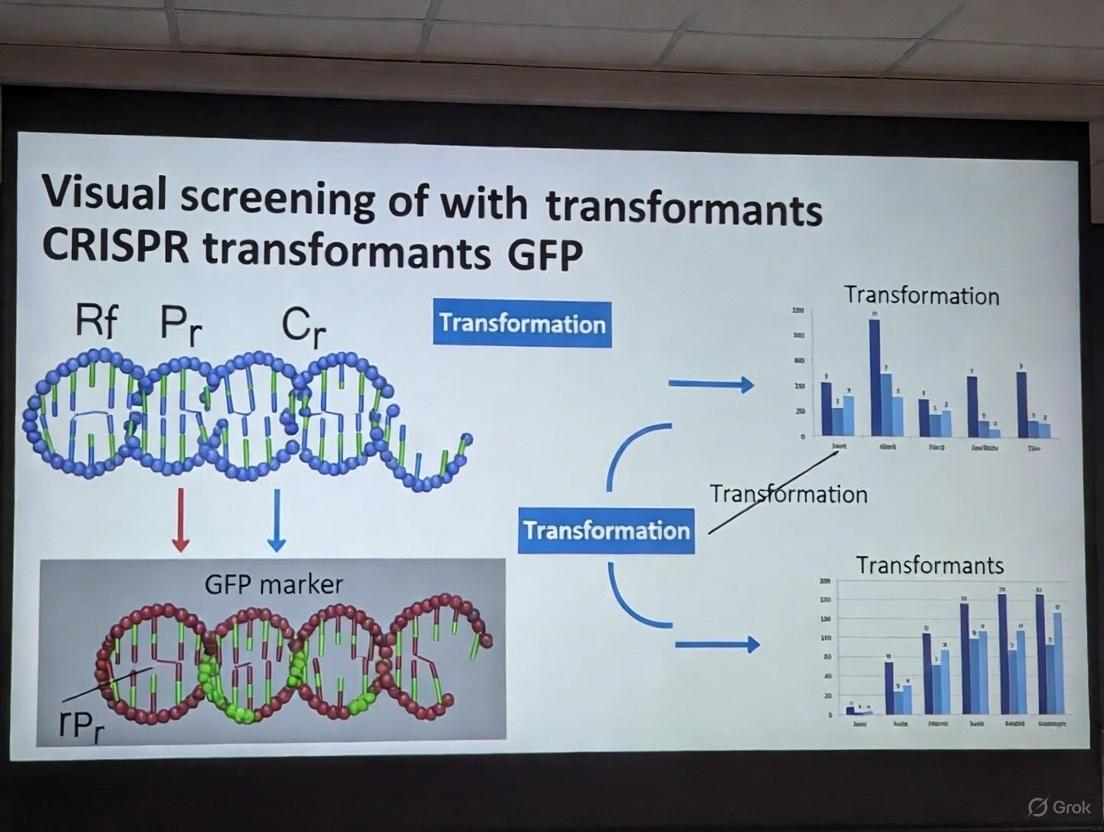
This article provides a comprehensive guide for researchers and drug development professionals on implementing visual screening of CRISPR transformants using GFP markers.
A Practical Guide to Visual CRISPR Screening: Using GFP Reporters for Efficient Transformat Selection and Analysis
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on implementing visual screening of CRISPR transformants using GFP markers. It covers the foundational principles of CRISPR-GFP reporter systems, from basic mechanisms to advanced screening setups. The content details practical methodologies for fluorescence-activated cell sorting (FACS)-based enrichment and high-throughput screening workflows. Critical troubleshooting sections address common challenges like low transfection efficiency and unexpected GFP expression. The guide also explores rigorous validation strategies to confirm editing outcomes, comparing GFP-based methods with other validation techniques. By synthesizing current best practices and emerging applications, this resource aims to enhance the efficiency and reliability of CRISPR screening campaigns in both basic research and therapeutic development.
GFP as a Visual Reporter in CRISPR Screening: Principles and System Design
The convergence of Green Fluorescent Protein (GFP) technology with CRISPR-Cas9 genome editing has revolutionized molecular biology, enabling real-time visual tracking of editing events within living cells. While GFP has been historically used as a quantitative reporter of gene expression [1], its adaptation into CRISPR systems provides researchers with a powerful tool to screen for successful transformants efficiently. These fluorescent CRISPR reporters function by linking a visible signal—the emission of green light—to the successful activity of the Cas9 nuclease or the precise incorporation of an edit via homology-directed repair. This direct visual feedback is invaluable for applications ranging from functional genomic screening to the development of cell and gene therapies, allowing scientists to bypass labor-intensive cloning and sequencing steps during initial screening phases. This article details the core mechanisms of these systems and provides standardized protocols for their implementation.
Core Mechanisms of CRISPR-GFP Reporter Systems
CRISPR-GFP reporters operate primarily through two ingenious molecular designs that couple the DNA cleavage or repair outcome to the functional expression of the fluorescent protein.
Frameshift-Based Detection of NHEJ
The most common mechanism involves the detection of Cas9-induced double-strand breaks that are repaired via the error-prone non-homologous end joining (NHEJ) pathway. In this setup, the coding sequence for a fluorescent protein like GFP or mCherry is cloned out-of-frame [2] [3]. Upstream of this fluorescent protein is a CRISPR target site—a copy of the genomic sequence that the sgRNA is designed to cut.
In the unedited state, the reporter is transcribed and translated, but due to the frameshift, the fluorescent protein is not produced, or only a non-functional peptide is made. When the Cas9/sgRNA complex successfully cleaves the reporter construct, the cellular NHEJ repair machinery introduces small insertions or deletions (indels) at the break site. A fraction of these indels will result in a frameshift mutation that places the fluorescent protein back into the correct reading frame. Consequently, the cell fluoresces, serving as a visual proxy for successful Cas9 cutting and NHEJ activity at the intended genomic locus [2] [3]. Systems like GEmCherry2 are engineered based on this principle, with optimizations such as the removal of alternative start codons to minimize background fluorescence [3].
HDR-Specific Reporter Systems
For applications requiring precise homology-directed repair (HDR), more sophisticated reporters like SRIRACCHA have been developed. This system uses a stably integrated reporter gene containing a puromycin resistance gene followed by the target site and an out-of-frame H2B-GFP reporter [3]. When a donor DNA template is co-transfected along with Cas9 and the sgRNA, a successful HDR event at the reporter locus uses the donor to correct the frame, leading to GFP expression. This HDR event in the reporter indicates that a parallel precise editing event is likely to have occurred at the endogenous genomic target [3]. A key advantage of the SRIRACCHA system is its reversibility, allowing for the removal of the reporter cassette after the desired mutant has been identified [3].
Table 1: Comparison of Key CRISPR-GFP Reporter Systems
| Reporter System | Core Mechanism | Repair Pathway Detected | Key Feature(s) | Primary Application |
|---|---|---|---|---|
| GEmCherry2 [3] | Out-of-frame mCherry | NHEJ | Low background; rapid sgRNA validation | Quantifying Cas9/sgRNA cutting efficiency |
| Dual Fluorochrome Reporter [2] | Out-of-frame GFP; iRFP transfection control | NHEJ | 17 target sites for multiplexing; enables enrichment of edited cells | Editing challenging cells (e.g., primary patient samples) |
| SRIRACCHA [3] | Out-of-frame H2B-GFP with donor template | HDR | Reversible integration; enriches for precise edits | Isolating cells with precise HDR-based genome edits |
The following diagram illustrates the logical workflow of the frameshift-based NHEJ reporter system:
Application Notes & Protocols
Protocol: Using a Frameshift GFP Reporter to Enrich KO Cells
This protocol is adapted from a study that successfully enriched CRISPR-edited patient-derived xenograft (PDX) cells, which are notoriously difficult to culture in vitro [2].
Key Materials:
- Cells: A cell line stably expressing Cas9 (e.g., NALM-6 used in the study).
- Reporter Plasmid: A dual-fluorochrome reporter construct (e.g., constitutively expressing iRFP-720 and an out-of-frame, destabilized GFP).
- sgRNA Vector: A lentiviral vector expressing your target sgRNA and a marker like mTagBFP.
Procedure:
- Stable Cell Line Generation: Lentivirally transduce your Cas9-expressing cells with the dual-fluorochrome reporter plasmid. Use flow cytometry to sort for iRFP-positive cells, establishing a polyclonal reporter cell line [2].
- Transduction with sgRNA: Transduce the reporter cell line with the lentiviral sgRNA vector. Aim for a low multiplicity of infection (MOI) to ensure single integrations and mimic conditions for challenging primary cells [2].
- Incubation and Expression: Culture the transduced cells for 3-5 days to allow for CRISPR cutting, repair, and GFP expression.
- Flow Cytometry and Sorting: Analyze the cells using a flow cytometer. First, gate for mTagBFP-positive cells (indicating sgRNA presence). Within this population, identify and sort the mTagBFP-iRFP-GFP triple-positive cells [2].
- Validation: The sorted GFP-positive population is highly enriched for cells with successful knockout (KO) at the genomic target. Validate editing efficiency via downstream methods like droplet digital PCR (ddPCR) or capillary immunoassay for protein loss [2].
Protocol: Rapid Generation of Homozygous Fluorescent Reporter Knock-In Pools
Generating knock-in reporter cell lines traditionally relies on tedious single-cell cloning. This protocol uses a single-plasmid system and FACS to rapidly create biallelic knock-in cell pools, preserving parental cell heterogeneity [4].
Key Materials:
- Single-Plasmid Construct: A vector combining the sgRNA (often under a doxycycline-inducible promoter) and the donor DNA template for HDR.
- Donor DNA Design: The donor should contain the fluorescent protein (e.g., GFP) sequence linked to the C-terminus of the endogenous target gene via a T2A "self-cleaving" peptide sequence. The start codon of the fluorescent reporter should be removed to prevent false positives from random integration [4].
Procedure:
- Electroporation: Deliver the single-plasmid construct into your target cells (e.g., mammalian cell lines like MEC or JHH5) using electroporation. The study used parameters of 870 V, 35 ms pulse width, and 2 pulses with the Neon Electroporation System [4].
- Induction and Culture: After electroporation, culture cells in medium containing 2 µg/mL doxycycline for 2 days to induce sgRNA expression. Then, replace with standard medium and culture for an additional ~10 days to allow for HDR and degradation of transiently expressed donor DNA [4].
- Fluorescence Detection and Sorting: After the culture period, use flow cytometry to detect and sort cells that are positive for the knock-in fluorescent reporter.
- Establishment of Stable Pools: Culture the sorted fluorescent cells to establish a stable knock-in cell pool. This pool will consist of a mixture of monoallelic and biallelic knock-in cells. The researchers noted that this method significantly reduces the rate of random integration compared to a dual-plasmid system [4].
Table 2: Troubleshooting Common Issues in CRISPR-GFP Reporter Assays
| Problem | Potential Cause | Suggested Solution |
|---|---|---|
| High background fluorescence | Alternative translation initiation; random integration of donor DNA. | Use optimized reporters like GEmCherry2 [3]; remove the start codon from the fluorescent reporter in the donor DNA [4]. |
| Low editing efficiency in GFP+ cells | Inefficient sgRNA; poor HDR efficiency. | Use the reporter to first validate and rank sgRNA efficiency [3]; use a single-plasmid system to improve HDR [4]. |
| Low signal-to-noise ratio in flow cytometry | Weak fluorescence from the reporter protein. | Use bright, stable fluorescent proteins like eYGFPuv [5] or link the GFP to a histone (H2B) for nuclear concentration [3]. |
| Poor enrichment of edited cells | Linker sequence issues leading to false negatives. | Incorporate a 48 bp glycine linker between the Cas9 target site and the GFP to prevent disruption of the GFP coding region during large deletions [2]. |
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of CRISPR-GFP reporter assays requires a suite of well-characterized reagents. The table below lists key materials and their functions.
Table 3: Essential Reagents for CRISPR-GFP Reporter Assays
| Reagent / Tool | Function | Examples & Notes |
|---|---|---|
| Fluorescent Reporter Plasmid | Provides the visual readout for editing. | GEmCherry2 (for NHEJ) [3]; SRIRACCHA (for HDR) [3]; Dual iRFP/GFP reporter [2]. |
| Cas9 Expression System | Provides the nuclease for DNA cleavage. | Stable cell line, transfected plasmid, or ribonucleoprotein (RNP) complexes. |
| sgRNA Expression Vector | Guides Cas9 to the specific genomic locus. | Can be cloned into vectors with fluorescent markers (e.g., mTagBFP) for tracking transduction [2]. |
| HDR Donor Template | Serves as a repair template for precise knock-in. | Designed with ~500-800 bp homology arms and a T2A-linked fluorescent protein without its start codon [4]. |
| Flow Cytometer / Cell Sorter | Essential for quantifying and isolating fluorescent cells. | Used for both analyzing editing efficiency and enriching positive populations [2] [4]. |
| Integrase-Deficient Lentivirus (IDLV) | Delivery method for transient expression of editing components without genomic integration. | Minimizes random integration risks; ideal for hard-to-transfect cells [4]. |
Within CRISPR-Cas9 genome editing, the efficient screening and isolation of successfully transformed cells is a critical step. Green Fluorescent Protein (GFP) reporter systems serve as a powerful tool for this visual screening, enabling researchers to rapidly identify edited cells. A fundamental design choice in developing these systems lies in the configuration of the GFP cassette: whether to use a promoter-driven or a promoterless construct. This article details the design considerations, experimental protocols, and key applications for both systems, providing a framework for their use in the visual screening of CRISPR transformants.
The core distinction between these systems hinges on the presence or absence of a dedicated promoter sequence upstream of the GFP gene. This choice dictates the experimental workflow, the interpretation of results, and the types of biological questions that can be addressed.
The logical relationship and primary applications of these two systems are summarized in the following diagram:
Promoter-Driven GFP Systems
In this conventional approach, the GFP gene is placed under the control of a strong, constitutive promoter (e.g., CMV, EF1α, or 35S in plants). This design ensures robust, continuous expression of GFP in any cell that has successfully incorporated the transgene, independent of the genomic integration site or the status of the target gene.
- Advantages: The system provides a bright, easily detectable signal that facilitates the rapid screening of positive transformants. It is particularly useful for tracking transfection efficiency and ensuring the presence of the CRISPR construct in cells [6].
- Disadvantages: A significant drawback is that GFP expression confirms only the presence of the vector, not successful on-target gene editing. Furthermore, the persistent presence of the transgene, including the fluorescent marker, can be undesirable for downstream applications or clinical use. A promoter-driven system can also lead to aberrant expression, as suggested by studies showing unexpected GFP expression even in the absence of a canonical promoter [7].
Promoterless GFP Systems
Promoterless designs place the GFP coding sequence without an upstream promoter. Expression is typically made dependent on a specific genomic event, such as successful Homology-Directed Repair (HDR) that places GFP in-frame with an endogenous, active promoter.
- Advantages: This system directly reports on a successful editing outcome. It enables the precise knock-in of a reporter and allows for the enrichment of cells that have undergone the desired HDR event, thereby reducing false positives from random integration [2] [4]. It is also instrumental in studying the activity of endogenous promoters.
- Disadvantages: The signal can be weaker and more variable, as it is subject to the regulation of the endogenous promoter. The design and construction are more complex, requiring careful consideration of homology arms and in-frame fusion. There is also evidence that GFP may exhibit low-level, aberrant expression even without a promoter, which could contribute to background noise [7].
Quantitative Performance Comparison
The choice between promoterless and promoter-driven systems involves trade-offs in editing efficiency, signal strength, and false-positive rates. The following table summarizes key performance metrics from published studies.
Table 1: Performance Comparison of Promoterless and Promoter-Driven GFP Reporter Systems
| System Type | Reported Editing Efficiency | Key Functional Outcome | False Positive/Background Signal Considerations |
|---|---|---|---|
| Promoter-Driven | 75-90% (Transient) [6] | Visual confirmation of transfection/transduction; Isolation of transgene-free mutants in subsequent generations [6]. | Potential for aberrant expression without promoter; confirms vector presence, not editing [7]. |
| Promoterless (HDR-dependent) | Up to 80% enrichment of edited alleles [2] | Successful knock-in and reporting on endogenous gene activity; Effective enrichment of HDR-edited cells [2] [4]. | Lower random integration; requires specific frameshift for activation in surrogate assays [2]. |
Experimental Protocols
Protocol: Using a Promoter-Driven GFP System to Identify CRISPR Transformants
This protocol is adapted from applications in plant and mammalian systems [6].
1. Materials:
- CRISPR/Cas9 vector with a constitutive promoter (e.g., 35S, CMV) driving GFP expression.
- Target cells (e.g., Arabidopsis, strawberry, soybean, or mammalian cell lines).
- Transformation/transfection reagents (e.g., Agrobacterium for plants, electroporation/lipofection for mammalian cells).
- Fluorescence microscope or flow cytometer.
2. Procedure:
- Step 1: Delivery. Introduce the CRISPR/Cas9-GFP vector into your target cells using the standard method for your system.
- Step 2: Screening (T0). After an appropriate incubation period (e.g., 2-7 days), screen cells or tissues for GFP fluorescence. GFP-positive events indicate successful delivery of the vector.
- Step 3: Mutation Analysis. Genotype the GFP-positive transformants using PCR/restriction enzyme digestion or sequencing to confirm on-target mutations at the genomic locus of interest.
- Step 4: Isolating Transgene-Free Mutants (T1/T2). Allow the primary transformants (T0) to self-cross or propagate. In the next generation (T1/T2), screen for individuals that show the expected mutant phenotype but lack GFP fluorescence. These are potential transgene-free mutants, as the CRISPR cassette has segregated away [6].
Protocol: Using a Promoterless GFP System to Enrich HDR-Edited Cells
This protocol is based on a dual-fluorochrome surrogate reporter system used in patient-derived xenograft (PDX) leukemia cells [2] [4].
1. Materials:
- Reporter Vector: Construct containing a constitutive red fluorescent protein (e.g., iRFP720) and an out-of-frame, promoterless GFP. The target sgRNA sequence is cloned upstream of GFP.
- CRISPR Components: Cas9 and sgRNA expression constructs.
- Target cells (e.g., NALM-6 cell line or primary PDX cells).
- Flow cytometer for cell sorting (FACS).
2. Procedure:
- Step 1: Co-transfection/Transduction. Deliver the promoterless GFP reporter vector along with the Cas9 and sgRNA constructs into the target cells.
- Step 2: Enrichment and Analysis. After 48-72 hours, analyze the cells by flow cytometry.
- First, gate for red fluorescent (iRFP720+) cells to select a population that has successfully taken up the reporter construct.
- Within this population, identify and sort the double-positive (iRFP720+/GFP+) cells. The expression of GFP indicates that a frameshift mutation has occurred at the target site, restoring the GFP reading frame and serving as a surrogate for Cas9 activity [2].
- Step 3: Validation. Validate the sorted cells by genotyping the endogenous target locus (e.g., via droplet digital PCR) to confirm a high frequency of indels. Protein analysis (e.g., Western blot) can confirm knockout of the target gene [2].
The workflow for this promoterless enrichment system is illustrated below:
The Scientist's Toolkit: Research Reagent Solutions
The following table lists essential reagents and their functions for implementing the described GFP reporter systems.
Table 2: Key Research Reagents for GFP Reporter Systems
| Reagent / Tool | Function in Reporter System | Example Use-Case |
|---|---|---|
| Constitutive Promoters (e.g., CMV, EF1α, 35S) | Drives strong, ubiquitous expression of GFP for tracking vector presence. | Visual screening of positive transformants in T0 generation [6]. |
| Dual-Fluorochrome Surrogate Reporter | Combines a constitutive marker (iRFP) with an out-of-frame GFP to enrich for nuclease-active cells. | Enriching CRISPR-edited PDX cells where HDR efficiency is low [2]. |
| T2A Self-Cleaving Peptide | Enables the co-translation of a gene of interest and GFP from a single transcript, often used in promoterless knock-in strategies. | Creating precise fluorescent reporter knock-in cell pools without a start codon on GFP [4]. |
| Integrase-Deficient Lentiviral Vector (IDLV) | Delivers transgenes transiently without genomic integration, minimizing random insertion. | Delivering CRISPR/sgRNA/donor DNA for HDR with reduced background [4]. |
| Native Visual Screening Reporter (NVSR) | Uses endogenous genes (e.g., FveMYB10 for anthocyanin) as a visible marker instead of GFP. | Identifying transgenic lines in plants without foreign fluorescent protein genes [8]. |
Both promoterless and promoter-driven GFP reporter systems are invaluable for the visual screening of CRISPR transformants, yet they serve distinct purposes. The promoter-driven approach offers a straightforward method for confirming transfection and initial transformation, and is highly effective for subsequently isolating transgene-free edited lines. In contrast, the promoterless strategy provides a more direct functional readout, enabling the precise enrichment of cells that have undergone the desired genome editing event, such as HDR, while minimizing false positives from random integration. The optimal design is dictated by the specific experimental goals, whether that is maximizing throughput and simplicity or ensuring precision and accurate reporting of endogenous gene activity.
In visual screening of CRISPR transformants using GFP markers, the selection of the genomic integration site is a fundamental determinant of success. A well-chosen locus ensures consistent, high-level expression of the GFP reporter, enabling reliable detection and selection of successfully edited cells without disrupting essential cellular functions. Targeting high-expression "safe harbor" loci, such as the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, provides a strategic solution to the common challenges of variable transgene expression and unpredictable phenotypic effects associated with random integration. This protocol details the rationale and methods for identifying and utilizing these optimal sites, with GAPDH serving as a primary model, to enhance the efficiency and reliability of CRISPR-based visual screening workflows.
Rationale for High-Expression Loci
Defining a Safe Harbor Locus for Visual Screening
A ideal locus for visual reporter integration exhibits several key characteristics:
- High and Stable Expression: The locus should reside within a transcriptionally active genomic region to drive sufficient GFP expression for easy detection across all transfected cells, minimizing false negatives.
- Neutrality for Cell Fitness: Integration at the site should not disrupt the function of essential genes or critical regulatory elements, thereby avoiding deleterious effects on cell growth, metabolism, or phenotype, which could confound experimental results.
- Open Chromatin Architecture: A chromatin environment that is more accessible to transcription machinery favors consistent and robust transgene expression.
The GAPDH locus exemplifies these properties. As a classic housekeeping gene constitutively expressed at high levels throughout the cell cycle, it provides a powerful endogenous promoter for driving GFP expression [9]. Furthermore, research has demonstrated that the precise integration of a transgene into the GAPDH locus via CRISPR/Cas9-mediated homologous recombination can be achieved without impairing the expression of the endogenous GAPDH gene itself, confirming its status as a safe harbor [9].
Established Safe Harbor Loci for CRISPR/GFP Work
While GAPDH is a robust choice, researchers have successfully targeted other loci for stable transgene expression. The table below summarizes several validated safe harbor loci.
Table 1: Established Genomic Safe Harbor Loci for Transgene Integration
| Locus Name | Organism | Key Characteristics | Application in CRISPR/GFP |
|---|---|---|---|
| GAPDH | Pig, Human, Mouse | High-expression housekeeping gene; integration shown not to affect endogenous gene expression [9]. | Knock-in of promoterless GFP cassettes; reliable visual marker detection. |
| Rosa26 | Mouse, Pig | Ubiquitously expressed genomic locus with high transcriptional activity; widely validated as a safe harbor [9]. | A standard site for landing various transgenes, including GFP reporters. |
| pH11 | Pig | Locus supports integration and expression of large transgenes (>9 kb) [9]. | Suitable for complex expression cassettes requiring high expression. |
| AAVS1 | Human | Safe harbor locus on human chromosome 19; known for open chromatin structure. | Common target for human cell line engineering with fluorescent reporters. |
Quantitative Data on Locus Performance
The effectiveness of a target locus is ultimately quantified by editing efficiency and reporter expression strength. The following table consolidates key performance metrics from published studies.
Table 2: Quantitative Performance Metrics of Selected Loci and Enhancement Strategies
| Parameter | Locus/System | Performance Metric | Experimental Context |
|---|---|---|---|
| Knock-in Efficiency | GAPDH Locus | Successful GFP knock-in and expression confirmed [9]. | Porcine fetal fibroblasts (PFFs). |
| Editing Enhancement | CRISPR/Cas9-RAD51 | ~2.5-fold increase in knock-out efficiency vs. standard CRISPR/Cas9 [10]. | HEK293T cells (targeting GAPDH). |
| System Efficiency | Plasmid-based (EPIC) | Average of 41.9% correct transformants [11]. | Candida auris protoplasts. |
| Editing Validation | GFP-to-RFP Conversion | >95% GFP-negative population indicating highly efficient Cas9 cleavage [12]. | Human gastric organoids (TP53/APC DKO). |
Experimental Protocols
Protocol 1: GFP Knock-in at the GAPDH Locus in Porcine Cells
This protocol is adapted from a study demonstrating the use of the GAPDH locus as a safe harbor for foreign gene knock-ins [9].
I. Research Reagent Solutions Table 3: Essential Reagents for GAPDH GFP Knock-in
| Reagent | Function/Description |
|---|---|
| PX330 Plasmid | CRISPR/Cas9 vector for expressing sgRNA and Cas9 nuclease [9]. |
| GAPDH-gRNA Oligos | Oligonucleotides encoding the sgRNA targeting the GAPDH locus. |
| pCDNA3.1-GAPDH-GFP-KI-donor | Donor vector containing a promoterless 2A-GFP cassette flanked by ~900 bp homology arms for the GAPDH locus [9]. |
| Lipofectamine 2000 | Transfection reagent for delivering plasmids into PK15 and 3D4/21 cell lines. |
| Electroporation Buffer | For transfection of primary porcine fetal fibroblasts (PFFs). |
| G418 (Geneticin) | Selective antibiotic for enriching transfected cells. |
II. Step-by-Step Workflow
- sgRNA Design and Cloning: Design an sgRNA to target the 3' end of the porcine GAPDH coding sequence, just before the stop codon. Anneal the oligonucleotides and ligate them into the BbsI site of the PX330 vector [9].
- Donor Vector Construction: Construct a donor plasmid containing a promoterless 2A-GFP sequence. This cassette must be flanked by homology arms (900 bp recommended) that are homologous to the sequences immediately upstream and downstream of the GAPDH target site. This design ensures that GFP is expressed only upon correct in-frame integration, driven by the endogenous GAPDH promoter [9].
- Cell Transfection:
- For PK15 and 3D4/21 cell lines: Co-transfect the PX330-GAPDH-sgRNA plasmid and the donor vector at a 1:1 ratio using Lipofectamine 2000.
- For Primary PFFs: Use electroporation (175 V/20 ms) to co-deliver the plasmids.
- Selection and Expansion: At 48 hours post-transfection, add G418 (700 µg/mL) to the culture medium for 3 days to select for cells that have incorporated the donor vector.
- Fluorescence-Activated Cell Sorting (FACS): Harvest the selected cells and use FACS to isolate a pure population of GFP-positive cells. These cells can then be expanded for further analysis [9].
- Validation:
- Immunofluorescence (IF): Confirm the co-localization of GFP and GAPDH signals using specific antibodies.
- Western Blot: Analyze protein lysates from sorted cells with anti-GFP and anti-GAPDH antibodies to verify the expression of both the endogenous GAPDH and the GFP-fusion protein [9].
Protocol 2: Enhancing CRISPR/GFP Workflow Efficiency with RAD51
To overcome variable editing efficiencies, particularly for knock-in strategies, co-expression of the homologous recombination protein RAD51 can be highly beneficial. This protocol is adapted from a study showing elevated CRISPR/Cas9-mediated genome editing efficiency with exogenous RAD51 [10].
I. Research Reagent Solutions Table 4: Essential Reagents for RAD51-Enhanced Editing
| Reagent | Function/Description |
|---|---|
| lentiCRISPR-RAD51-GFP Plasmid | An all-in-one vector constitutively expressing Cas9, a specific sgRNA, and RAD51 via 2A peptides, along with a puromycin resistance marker [10]. |
| Puromycin | Selective antibiotic for cells containing the CRISPR plasmid. |
| T7 Endonuclease I (T7E1) | Enzyme for detecting indel mutations and assessing cutting efficiency. |
II. Step-by-Step Workflow
- Vector Construction: Generate an all-in-one CRISPR/Cas9-RAD51 plasmid. This involves cloning an E2A-RAD51-T2A-EGFP cassette into a lentiCRISPR vector, downstream of the Cas9 sequence, creating a quad-cistronic expression system (Cas9-RAD51-EGFP-PuroR) [10].
- Cell Transfection and Selection: Transfect the construct into your target cell line (e.g., HEK293T). After 48 hours, replace the medium and add puromycin to select for successfully transfected cells for 72 hours.
- Efficiency Analysis:
- T7E1 Assay: Harvest genomic DNA from selected cells. Amplify the genomic region surrounding the CRISPR target site by PCR. Denature and reanneal the PCR products to form heteroduplexes. Treat with T7E1 enzyme and analyze the cleavage products by gel electrophoresis. A higher proportion of cleaved bands indicates a higher rate of indel formation, signifying improved editing efficiency [10].
- Sequencing: Clone the PCR products and perform Sanger sequencing on multiple colonies to precisely quantify the editing efficiency and the spectrum of induced mutations [10].
Molecular Mechanism of Targeted Integration
The following diagram illustrates the key molecular steps that occur during homology-directed repair (HDR) for precise GFP cassette integration into a target locus like GAPDH, and how RAD51 enhances this process.
The strategic selection of high-expression, phenotypically neutral loci such as GAPDH is a critical factor for the success of visual screening in CRISPR experiments. The protocols outlined here provide a reliable framework for achieving efficient GFP reporter knock-in and robust expression. Furthermore, the integration of enhancing strategies, like RAD51 co-expression, can significantly increase editing efficiency, reducing screening effort and time. By adopting these targeted approaches, researchers can generate more consistent and interpretable data, thereby accelerating discoveries in functional genomics and drug development.
The efficacy of CRISPR-Cas9 genome editing is fundamentally dependent on the coordinated delivery and performance of its two core components: the Cas nuclease and the guide RNA (gRNA). Achieving high editing efficiency requires a careful balance between sufficient Cas9 expression and the use of highly efficient, specific gRNAs. This balance is particularly critical in experiments involving visual screening with fluorescent markers like GFP, where editing outcomes must be accurately and efficiently tracked. This protocol provides detailed methodologies for optimizing CRISPR component delivery and validation, with specific application to visual screening systems. We present optimized parameters for achieving high knockout efficiencies across single and multiple genes, quantitative frameworks for gRNA selection, and practical tools for implementation in research settings.
Core Optimization Strategies
Establishing a High-Efficiency Cas9 Expression System
A doxycycline-inducible spCas9-expressing human pluripotent stem cell (hPSC-iCas9) system provides tunable nuclease expression with significant advantages over constitutive systems. Through systematic optimization of critical parameters, this system achieved remarkable efficiency: 82–93% stable INDELs (Insertions and Deletions) for single-gene knockouts, over 80% for double-gene knockouts, and up to 37.5% homozygous knockout efficiency for large DNA fragment deletions [13].
Key optimized parameters in the hPSC-iCas9 system include:
- Cell tolerance to nucleofection stress: Pre-optimization of cell viability post-electroporation
- Transfection methodology: Use of chemical synthesized and modified sgRNA (CSM-sgRNA) with 2’-O-methyl-3'-thiophosphonoacetate modifications at both 5’ and 3’ ends to enhance sgRNA stability
- Nucleofection frequency: Implementation of repeated nucleofection 3 days after initial transfection
- Cell-to-sgRNA ratio: Optimization of the relationship between cell numbers and sgRNA quantities [13]
gRNA Design and Validation Framework
gRNA design critically impacts editing efficiency. Experimental validation of three widely used gRNA scoring algorithms demonstrated that Benchling provided the most accurate predictions for sgRNA efficiency [13]. However, algorithm predictions alone are insufficient, as evidenced by the discovery of an ineffective sgRNA targeting exon 2 of ACE2 that exhibited 80% INDELs but retained ACE2 protein expression [13]. This highlights the necessity of experimental validation through Western blotting or functional assays to confirm protein knockout rather than relying solely on INDEL frequency.
Table 1: Comparison of gRNA Design and Optimization Approaches
| Approach | Key Features | Efficiency Outcomes | Validation Requirements |
|---|---|---|---|
| In vitro transcribed sgRNA (IVT-sgRNA) | Standard transcription method | Variable efficiency; subject to degradation | INDEL analysis via T7EI assay or sequencing |
| Chemical synthesized modified sgRNA (CSM-sgRNA) | 2’-O-methyl-3'-thiophosphonoacetate modifications at 5'/3' ends | Enhanced stability and editing efficiency | Protein-level validation (Western blot) |
| Dual-gRNA approach | Two gRNAs targeting same gene or adjacent loci | Up to 80% efficiency for double knockouts; large fragment deletion | PCR confirmation of deletion size |
| Algorithm-predicted gRNAs | Benchling, CCTop, or other prediction tools | Varies by algorithm accuracy | Multi-level validation (INDEL + protein) |
Experimental Protocols
Protocol: Rapid Screening of CRISPR Editing Outcomes Using Fluorescent Protein Conversion
This protocol adapts a system for distinguishing DNA damage repair outcomes by converting enhanced green fluorescent protein (eGFP) to blue fluorescent protein (BFP) through targeted mutation, enabling rapid assessment of gene knockout efficiency [14].
Materials
- eGFP-positive cell line
- CRISPR-Cas9 components (Cas9 + gRNAs targeting eGFP)
- Tissue culture equipment and reagents
- Flow cytometer with appropriate filters for eGFP and BFP
- Nucleofection system or transfection reagent
Methodology
Day 1: Cell Preparation
- Culture eGFP-positive cells to 80-90% confluency in appropriate medium.
- Dissociate cells using 0.5 mM EDTA or appropriate dissociation reagent.
- Count cells and resuspend at desired concentration for transfection.
Day 2: Transfection
- Prepare transfection complexes:
- For RNP delivery: Complex 5μg of purified Cas9 protein with 2μg of sgRNA targeting eGFP (sequence designed to convert eGFP to BFP) for 10 minutes at room temperature.
- For plasmid delivery: Use 2-5μg of plasmid expressing both Cas9 and sgRNA.
- Transfect using optimized method (nucleofection program CA137 for hPSCs or lipid-based transfection for other cell types).
- Plate transfected cells in appropriate growth medium.
Day 3-6: Expression and Analysis
- Allow cells to recover and express edited phenotypes for 72-96 hours.
- Analyze cells using flow cytometry to detect eGFP loss and BFP gain.
- Calculate editing efficiency as percentage of BFP-positive cells or eGFP-negative cells.
- Isolate successfully edited populations using fluorescence-activated cell sorting (FACS) if needed [14].
Protocol: Optimized Dual-gRNA Delivery for Enhanced Knockout Efficiency
This protocol describes an optimized approach for delivering two gRNAs simultaneously to enhance knockout efficiency, using FolicPolySpermine nanoparticles as a delivery vehicle [15].
Materials
- FolicPolySpermine nanoparticles (spermine, polyethylene glycol, and folic acid-based)
- Two gRNAs targeting adjacent sites in the gene of interest
- Cas9 expression plasmid or Cas9 protein
- HEK293T cells or other relevant cell line
- Restriction enzymes (FastDigest BpiI) for cloning
- PCR amplification and sequencing reagents
Methodology
Step 1: gRNA Design and Cloning
- Design two gRNAs targeting adjacent regions (within 2.2 kb) of the target gene using http://crispr.mit.edu/ or Benchling.
- Synthesize sense and antisense oligonucleotides for each gRNA, hybridize to form duplexes.
- Clone each duplex into the restriction site of FastDigest BpiI in the PX458 (Addgene #48138) expression plasmid using standard molecular biology techniques.
- Verify successful cloning by colony PCR and direct sequencing.
Step 2: Nanoparticle Preparation and Transfection
- Prepare FolicPolySpermine nanoparticles according to established protocols [15].
- Complex nanoparticles with CRISPR plasmids (2-5μg total DNA) at optimal weight ratios.
- Transfect HEK293T cells (or target cell line) using nanoparticle complexes.
- Include controls transfected with lipofectamine 2000 for comparison.
Step 3: Validation and Analysis
- Harvest cells 72-96 hours post-transfection.
- Extract genomic DNA and perform PCR amplification across the target region.
- Analyze deletion efficiency via gel electrophoresis (size shift) and Sanger sequencing.
- Confirm protein knockout via Western blotting if antibodies are available [15].
Quantitative Framework for gRNA Selection
The following table summarizes key efficiency metrics for different CRISPR delivery and gRNA selection approaches, enabling evidence-based experimental design:
Table 2: Efficiency Metrics for CRISPR Delivery and gRNA Selection Approaches
| Parameter | High-Efficiency Benchmark | Key Factors Influencing Efficiency | Validation Method |
|---|---|---|---|
| Single-gene knockout | 82-93% INDELs [13] | gRNA efficiency, Cas9 delivery method, cell type | ICE analysis, TIDE |
| Dual-gene knockout | >80% efficiency [13] | gRNA pairing, distance between targets | PCR, sequencing |
| Large fragment deletion | Up to 37.5% homozygous knockout [13] | Distance between gRNAs (up to 2.2 kb) | PCR product size analysis |
| gRNA prediction accuracy | Benchling most accurate [13] | Algorithm selection, on-target score | Experimental validation |
| Ineffective gRNA detection | 80% INDELs with protein retention [13] | Reading frame shifts, protein domains | Western blot |
Visual Workflows for Experimental Planning
The following diagrams illustrate key experimental workflows and relationships in CRISPR component delivery and screening.
Diagram 1: CRISPR Workflow with Fluorescent Screening
Diagram 2: gRNA Efficiency Balance
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Component Delivery and Screening
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| Cas9 Expression Systems | Doxycycline-inducible spCas9 hPSCs | Tunable Cas9 expression; reduces cytotoxicity |
| gRNA Synthesis | Chemical synthesized modified sgRNA (CSM-sgRNA) | Enhanced nuclease resistance; improved stability |
| Delivery Vehicles | FolicPolySpermine nanoparticles | Targeted, efficient CRISPR plasmid delivery [15] |
| Delivery Vehicles | Lipid Nanoparticles (LNPs) | High-efficiency in vivo delivery; suitable for redosing [16] |
| Fluorescent Reporters | eGFP-to-BFP conversion system | Rapid visual screening of editing outcomes [14] |
| Analysis Algorithms | ICE (Inference of CRISPR Edits) | Accurate INDEL quantification from sequencing data [13] |
| Analysis Algorithms | Benchling gRNA designer | Predictive scoring of gRNA efficiency [13] |
| Cloning Systems | PX458 (Addgene #48138) | All-in-one Cas9 and gRNA expression vector [15] |
Within the field of molecular biology, particularly in the visual screening of CRISPR transformants, Green Fluorescent Protein (GFP) has established itself as a pivotal reporter tool. This application note delineates the specific scenarios where GFP-based screening offers significant advantages over traditional selection methods, such as antibiotic resistance or phenotypic assays. We detail the quantitative performance metrics of GFP screening, provide a comprehensive protocol for its implementation in CRISPR/SpCas9 workflows, and visualize the core methodology. By synthesizing current research, we aim to equip researchers with the knowledge to effectively apply GFP screening to accelerate the isolation of genetically modified cells.
The advent of fluorescent proteins has revolutionized the tracking of gene expression and the selection of engineered cells. While traditional methods often rely on antibiotic selection, which confirms the presence of a resistance marker but not the functional expression of the cargo gene, GFP screening provides a direct, visual, and often quantitative readout of successful genetic modification [1]. In the specific context of CRISPR/Cas9 research, this allows researchers to directly screen for cells that are not only transformed but are also actively expressing the Cas9 machinery, thereby increasing the likelihood of successful editing [17]. However, the technique is not without its limitations, including potential interference with Cas9 activity and challenges in identifying Cas9-free edited progeny. This document explores the balance of these advantages and limitations, providing a framework for researchers to determine when GFP screening is the most effective tool.
Key Advantages and Documented Superiority of GFP Screening
GFP screening provides several distinct advantages that make it superior to traditional methods in many experimental contexts.
Direct, Rapid, and Non-Destructive Visualization
The most significant advantage of GFP is the ability to visually identify positive transformants in real-time without harming the cells. This non-destructive quality allows for the tracking of gene expression kinetics and the easy isolation of live, positive cells for further expansion and analysis. A 2025 study on plant CRISPR systems directly contrasted this with antibiotic selection, finding that screening with GFP or RNA aptamers provided a more direct method for identifying positive T1 transformants than selection with hygromycin resistance alone [17].
Quantitative and High-Throughput Capabilities
GFP serves not just as a qualitative marker but also as a quantitative reporter of gene expression. Flow cytometric measurement of GFP fluorescent intensity has been shown to be directly proportional to both GFP mRNA abundance and the underlying gene copy number, enabling precise assessment of promoter activity [1]. This facilitates high-throughput screening, as demonstrated by automated systems like the QPix 400 series, which can pick over 3,000 colonies per hour based on user-defined fluorescence intensity thresholds, a five-fold increase over manual picking [18].
Enabling Complex Functional Assays
GFP-based readouts can be engineered into sophisticated assays that go beyond simple transformation. For instance, the FAST (Fluorescent Assembly of Split-GFP for Translation Tests) method uses the complementation of GFP1-10 and GFP11 fragments to detect cell-free protein synthesis with a sensitivity of 8 ± 2 pmol of polypeptide, a use case where traditional radioactive labeling would be hazardous and complex [19]. Similarly, split-GFP systems have been adapted to quantify the display of proteins on the microbial cell surface, a task difficult to accomplish with conventional immunoassays that require costly and time-consuming antibody generation [20].
Inherent Limitations and Challenges
Despite its power, GFP screening is not a universal solution and possesses several key limitations that researchers must consider.
Technical Complexity and False Signals
A primary challenge is the potential for false positives and negatives. In the plant CRISPR study, the conventional GFP/Cas9 system had a 40% omission rate, failing to identify many positive transformants that were detected via genomic PCR. This was attributed to the incomplete cleavage of the 2A peptide linking GFP to Cas9, which can impair Cas9 activity and reduce fluorescence [17]. Furthermore, fluorescence can be detected if the fluorescent protein is retained in the cytoplasm, obscuring accurate localization in surface display experiments [20].
Potential for Interference and Stability Issues
The relatively large size of GFP (∼25 kDa) can potentially interfere with the function, folding, or localization of the protein it is fused to. This has spurred the development of smaller RNA aptamer reporters as alternatives [17]. Additionally, GFP fluorescence is dependent on chromophore maturation, which has a slow rate compared to protein folding kinetics, potentially delaying the readout [19]. GFP fluorescence can also be disrupted by certain small-molecule drugs, such as the covalent kinase inhibitors osimertinib, afatinib, and neratinib, which can confound results in drug screening assays [21].
Inability to Directly Isolate Cas9-Free Progeny
In CRISPR workflows, a critical goal is to identify edited organisms that have segregated away from the Cas9 transgene. A GFP signal linked to Cas9 expression makes it impossible to distinguish between a Cas9-positive plant and a Cas9-free, edited plant in the T2 generation, necessitating additional molecular screening to confirm the loss of the transgene [17].
Table 1: Quantitative Comparison of GFP Screening vs. Traditional Selection in Documented Studies
| Experimental Context | GFP Screening Performance | Traditional Method Performance | Reference |
|---|---|---|---|
| CRISPR T1 Transformant Selection | 60% identification accuracy (40% omission rate) | Hygromycin resistance: 100% selection efficiency but includes escapes | [17] |
| Bacterial Colony Picking | >3,000 colonies/hour; selection based on fluorescence intensity | ~600 colonies/hour manually; selection based on visual phenotype | [18] |
| Cell-Free Protein Synthesis | Sensitivity: 8 ± 2 pmol of polypeptide; non-hazardous | Radioactive labeling: hazardous, technically complex, time-consuming | [19] |
| Microbial Surface Display | One-step, no antibody cost; quantitative via flow cytometry | Immunoassays: costly antibodies, multiple washing steps, hours to complete | [20] |
Application Note: GFP Screening in a CRISPR/SpCas9 Workflow
The following protocol is adapted from a 2025 study that developed an RNA aptamer-assisted CRISPR/Cas9 system, with steps relevant to GFP screening detailed for the isolation of positive Arabidopsis thaliana T1 transformants [17].
Experimental Workflow
The diagram below outlines the key steps for screening CRISPR transformants using a GFP reporter system.
Detailed Step-by-Step Protocol
Step 1: Vector Construction and Transformation
- Clone your gene of interest and the sgRNA(s) into a CRISPR vector where the Cas9 gene is fused in-frame to the GFP reporter gene via a self-cleaving 2A peptide (e.g., P2A, T2A).
- Introduce the constructed vector into your target organism. For Arabidopsis thaliana, use the Agrobacterium-mediated floral dip method [17].
Step 2: Primary Selection and Screening
- After transformation, harvest T1 seeds and plate on solid MS medium containing the appropriate antibiotic (e.g., hygromycin) to select for transformants.
- Grow seedlings under standard conditions for 7-10 days.
- Screen the antibiotic-resistant seedlings for GFP fluorescence using a fluorescent stereomicroscope with a standard GFP filter set (Ex/Em: ~488/507 nm).
- Note: The user-defined fluorescence threshold is critical. The QPix 400 system, for example, allows setting a minimum Mean Fluorescence Intensity (e.g., >40,000) for automated picking [18].
Step 3: Validation and Downstream Analysis
- Carefully collect the GFP-positive seedlings and transfer them to soil for expansion.
- Extract genomic DNA from leaf tissue of the expanded positives.
- Perform PCR amplification of the target genomic region and validate editing efficiency via sequencing (e.g., Sanger or NGS).
- Critical Limitation Check: Be aware that fluorescence indicates Cas9 expression, not necessarily successful editing. Furthermore, the 2A peptide linkage may be incomplete, leading to false negatives and reduced Cas9 activity [17]. Always confirm edits molecularly.
The Scientist's Toolkit: Essential Reagents and Solutions
Table 2: Key Research Reagents for GFP Screening in CRISPR Workflows
| Reagent / Solution | Function in Protocol | Example & Notes |
|---|---|---|
| CRISPR/GFP Vector | Expresses Cas9, sgRNA, and GFP reporter. | Plasmid with Cas9-P2A-GFP fusion; available from Addgene. P2A peptide allows co-translational cleavage. |
| Selection Antibiotic | Primary selection for transformants. | Hygromycin for plants; Ampicillin or Kanamycin for bacterial systems. |
| Fluorescence Microscope | Visualization and manual picking of GFP+ cells/colonies. | Requires standard GFP filter set (Ex ~488 nm, Em ~507 nm). |
| Automated Colony Picker | High-throughput, quantitative screening of colonies. | QPix 400 Series with fluorescence module; allows setting intensity thresholds [18]. |
| Split-GFP Components | For detecting protein expression, display, or interactions. | GFP1-10 (25 kDa) and GFP11 (16 aa) fragments; used in FAST and surface display assays [19] [20]. |
| Fluorescence-Compatible Plates | For quantitative assays in cell culture or liquid samples. | Black-walled, clear-bottom plates for reading in plate readers. |
GFP screening outperforms traditional selection methods by providing direct, quantitative, and non-destructive visualization of gene expression, which is invaluable for high-throughput workflows and functional assays in CRISPR research. Its primary advantages lie in speed, visual confirmation, and the rich quantitative data it provides. However, limitations such as the potential for false negatives due to fusion protein issues, the inability to screen for Cas9-free edits, and the size of the GFP protein itself necessitate a complementary approach. Researchers are advised to use GFP screening as a powerful first-pass filter but to always couple it with robust molecular validation techniques to confirm genuine genetic edits.
Implementing GFP-Based CRISPR Screens: From FACS to High-Throughput Workflows
In the field of functional genomics and drug discovery, CRISPR-Cas9 gene editing has revolutionized target identification and validation. A critical step in this process is the rapid and accurate assessment of DNA repair outcomes following CRISPR-induced DNA breaks. Fluorescence-based screening pipelines provide a powerful solution, enabling researchers to distinguish between different gene editing results efficiently and at scale [22].
This protocol details the establishment of a fluorescence-based screening pipeline using an enhanced Green Fluorescent Protein (eGFP) to Blue Fluorescent Protein (BFP) conversion system. The core principle leverages the fact that successful gene editing alters the fluorescent phenotype of cells, allowing for straightforward differentiation between various DNA repair outcomes. This method is particularly valuable for high-throughput assessment of gene editing techniques, which is crucial for pharmaceutical and biotechnology research [14].
The system is designed to distinguish between two primary DNA repair pathways:
- Non-homologous end joining (NHEJ), which typically results in gene knockout (loss of fluorescence).
- Homology-directed repair (HDR), which can lead to precise gene correction or mutation (shift from green to blue fluorescence) [14].
Key Research Reagent Solutions
The successful implementation of this screening pipeline relies on several crucial reagents and their specific functions, as outlined in the table below.
Table 1: Essential Research Reagents and Their Functions in the Fluorescence Screening Pipeline
| Reagent / Component | Function / Explanation |
|---|---|
| eGFP Reporter Cell Line | Provides the chromosomal target for CRISPR-Cas9 editing; successful editing alters its fluorescent signal [14]. |
| CRISPR-Cas9 System | RNA-guided endonuclease that creates a precise double-strand break in the DNA at the target eGFP locus [22]. |
| sgRNA Targeting eGFP | Directs the Cas9 nuclease to the specific sequence within the eGFP gene that is to be modified [14]. |
| HDR Donor Template | A DNA template containing the desired BFP mutation; used by the cell's repair machinery to convert eGFP to BFP [14]. |
| Fluorogenic Proteins (e.g., tdTomato-tDeg) | Engineered fluorescent proteins that become stable and fluorescent only upon binding to a specific RNA aptamer (e.g., Pepper), drastically reducing background noise in imaging applications [23]. |
| Pepper-fused sgRNA | A modified sgRNA that incorporates the Pepper RNA aptamer; it recruits and stabilizes the fluorogenic protein, enabling high-contrast imaging of genomic loci [23]. |
| dCas9 (Nuclease-deficient Cas9) | A catalytically "dead" Cas9 that can target genomic DNA without cutting it; serves as a platform for fluorogenic CRISPR (fCRISPR) imaging systems [23]. |
The following diagram illustrates the logical flow and key decision points of the fluorescence-based screening protocol, from cell preparation to final analysis.
Detailed Experimental Protocol
Protocol for Screening CRISPR-Cas9 Outcomes via eGFP to BFP Mutation
This protocol provides a step-by-step methodology for distinguishing between NHEJ-induced gene knockout and HDR-induced gene mutation in a cell population [14].
Table 2: Step-by-Step Protocol for Fluorescence-Based Screening
| Step | Procedure | Key Parameters | Purpose |
|---|---|---|---|
| 1. Cell Preparation | Culture and maintain eGFP-positive cells. Ensure high viability and optimal confluency (e.g., 70-80%) before transfection. | Cell line of choice, growth medium, cell viability >95%. | To provide a uniform, healthy population of cells expressing the target eGFP gene. |
| 2. Transfection | Co-transfect cells with plasmids encoding: a) Cas9 nuclease, b) sgRNA targeting the eGFP gene, and c) HDR donor template for BFP conversion. | Use optimized transfection reagent or method (e.g., lipofection, electroporation). Controls (e.g., sgRNA only) are essential. | To deliver the gene editing machinery and donor template into the cells to initiate the DNA break and repair process. |
| 3. Incubation & Expression | Incubate transfected cells for a sufficient period (e.g., 48-72 hours) to allow for DNA repair, expression, and maturation of the new fluorescent protein (BFP). | Standard cell culture conditions (37°C, 5% CO₂). BFP maturation time. | To enable the cellular repair mechanisms (NHEJ or HDR) to act and for the resulting fluorescent phenotypes to manifest. |
| 4. Analysis & Sorting | Analyze cells using flow cytometry or fluorescence microscopy. Measure fluorescence in eGFP and BFP channels. | Use appropriate laser/filter sets for eGFP (Ex/~488nm, Em/~510nm) and BFP (Ex/~405nm, Em/~450nm). | To identify and quantify the proportions of cells that have undergone successful HDR (BFP+), NHEJ (non-fluorescent), or no editing (eGFP+). |
| 5. Data Interpretation | Calculate editing efficiencies based on the population shifts in fluorescence. | % BFP+ cells = HDR efficiency; % non-fluorescent cells = NHEJ efficiency. | To quantitatively assess the outcomes and efficacy of the gene editing procedure. |
Advanced Imaging with Fluorogenic CRISPR (fCRISPR)
For high-contrast imaging of genomic loci, such as visualizing the site of CRISPR action, a fluorogenic CRISPR (fCRISPR) system is recommended. This method offers superior signal-to-noise ratio compared to conventional methods using constitutively fluorescent proteins [23].
System Design: The fCRISPR system employs three components:
- dCas9: Catalytically dead Cas9 for targeting without cutting.
- Modified sgRNA: The sgRNA scaffold is engineered with inserted Pepper RNA aptamers in the tetraloop and stem-loop 2.
- Fluorogenic Protein: A fluorescent protein (e.g., tdTomato) fused to an unstable degron domain (tDeg). This protein is rapidly degraded unless bound to the Pepper aptamer.
Imaging Procedure:
- Co-transfect cells with plasmids expressing the Pepper-fused sgRNA, dCas9, and the tdTomato-tDeg fluorogenic protein.
- The dCas9:sgRNA complex binds the target genomic DNA.
- The Pepper aptamer on the sgRNA recruits and binds the tdTomato-tDeg protein, concealing its degron. This binding stabilizes the protein and activates its fluorescence specifically at the target locus.
- Image the cells using standard fluorescence microscopy. The fCRISPR system produces bright, specific puncta against a very low background, as unbound fluorogenic proteins are degraded [23].
Data Analysis and Presentation
Effective presentation of quantitative data is crucial for interpreting screening results. The following table and descriptions outline standard methods for data summarization and visualization.
After analysis, data should be summarized by group (e.g., different experimental conditions or editing outcomes). A key numerical summary is the difference between means (or medians) of the compared groups [24].
Table 3: Generalized Structure for Presenting Quantitative Data from a Screening Experiment
| Experimental Group | Mean Editing Efficiency (%) | Standard Deviation | Sample Size (n) |
|---|---|---|---|
| Condition A | Value | Value | Value |
| Condition B | Value | Value | Value |
| Difference (A - B) | Value | -- | -- |
Data Visualization Techniques
Choosing the right graph is essential for comparing quantitative data across different groups [24]:
- Boxplots: The best general choice for displaying distributions and comparing medians, quartiles, and ranges between multiple groups. They readily show outliers and the overall spread of data [24].
- 2-D Dot Charts: Ideal for small to moderate amounts of data, as they show individual data points, preventing the loss of detail that can occur with summary graphics like boxplots. Points are often jittered or stacked to avoid overplotting [24].
- Back-to-Back Stemplots: A good option for small datasets when comparing only two groups, as they preserve the original data values [24].
Application in Drug Discovery
This fluorescence-based screening pipeline is highly applicable in pharmaceutical research. It can be used to:
- Identify and validate new biological targets for precision medicines through functional genomic screening [22].
- Understand drug resistance and sensitivity mechanisms ahead of clinical trials by performing positive selection screens for genes that confer resistance to a cytotoxic agent [22].
- Define gene essentiality in specific cancer cell lines, creating a landscape of gene dependency which can inform therapeutic strategies (a classic negative selection screen) [22].
Fluorescence-Activated Cell Sorting (FACS) has become an indispensable tool for modern biological research, particularly in the field of CRISPR-based genetic engineering. The ability to isolate specific cellular populations based on fluorescent markers such as Green Fluorescent Protein (GFP) enables researchers to study gene function, protein localization, and cellular responses with remarkable precision. Within the context of CRISPR transformant screening, FACS provides a powerful method for identifying successfully edited cells, characterizing editing efficiencies, and isolating transgene-free progeny for downstream applications. This application note details established protocols and strategic considerations for effective FACS-based enrichment of GFP-positive and GFP-negative populations, with a specific focus on applications in CRISPR-Cas9 visual screening. The integration of these techniques is crucial for advancing functional genomics and accelerating the development of genetically engineered organisms for both basic research and therapeutic purposes.
Key Experimental Workflows
The following diagrams illustrate the core workflows for standard GFP-based sorting and for addressing the common challenge of cellular autofluorescence.
Core FACS Workflow for CRISPR Screening
Gating Strategy to Exclude Autofluorescence
Critical Reagents and Equipment
The success of FACS-based enrichment relies on a well-characterized set of reagents and instruments. The following toolkit outlines essential components.
Table: Research Reagent Solutions for FACS-Based GFP Sorting
| Reagent/Equipment | Function/Application | Specific Examples & Notes |
|---|---|---|
| GFP Reporter Construct | Visual marker for CRISPR delivery and success; co-expressed with Cas9 | Can be linked via 2A self-cleaving peptide or expressed from a separate promoter [17] |
| Cell Dissociation Reagent | Generation of high-quality single-cell suspension | Accutase is preferred over trypsin as it dislodges cells without damaging surface proteins [25] |
| Viability Stain | Discrimination and exclusion of dead cells | Propidium Iodide or DAPI; critical for improving sort purity and downstream cell health |
| FACS Buffer | Maintains cell viability and prevents clumping during sort | PBS + 1% FBS + 2.5 mM EDTA + 25 mM HEPES [26] [25] |
| Fluorescence-Activated Cell Sorter | Instrument for analyzing and physically separating cells | Standard commercial FACS machines (e.g., BD Influx) are suitable; no custom FADS required [27] [28] |
| Sort Collection Tubes | Receives sorted cell populations while maintaining sterility and viability | Tubes pre-filled with collection medium (e.g., PBS + 1% FBS or culture medium) [25] |
Comparative Performance of Enrichment Strategies
Different screening and enrichment strategies offer distinct advantages in terms of efficiency, accuracy, and applicability. The quantitative data below compares several approaches.
Table: Quantitative Comparison of Fluorescence-Based Sorting Strategies
| Method / System | Reported Enrichment Efficiency | Key Advantages | Primary Application Context |
|---|---|---|---|
| Conventional GFP/Cas9 | 40% omission rate in T1 transformant identification [17] | Established, widely used protocol | General CRISPR screening in plant and mammalian cells [17] |
| RNA Aptamer (3WJ-4×Bro/Cas9) | 78.6% increase in T1 mutation rate vs. GFP/Cas9; 30.2% improved Cas9-free mutant sorting [17] | Higher accuracy; avoids fluorescent protein interference with Cas9 activity | Plant genome editing, particularly for selecting transgene-free edited lines [17] |
| Autofluorescence-Restrictive Gating | Up to 7-fold enrichment of true eGFP+ cells vs. standard protocol [26] | Effectively excludes false positives from intrinsically autofluorescent cells (e.g., RPE) | Gene therapy assessment in hard-to-transduce, autofluorescent cell types [26] |
| MACS Pre-enrichment | Can increase target cell frequency >30-fold before FACS [29] | Higher cell yield (91-93% vs. ~30% for FACS); faster for multiple samples [28] | High-yield preliminary enrichment when ultimate purity is not required [28] [29] |
Detailed Experimental Protocols
Basic Protocol: Standard FACS for GFP+ CRISPR Transformants
This protocol is adapted from established methods for sorting live mammalian cells based on surface and intracellular markers [25].
Materials:
- Library of mutant cells (e.g., CRISPR-Cas9 transfected)
- Fetal Bovine Serum (FBS)
- Phosphate-Buffered Saline (PBS, without Ca²⁺ or Mg²⁺)
- Accutase cell detachment solution
- FACS Buffer: PBS + 1% FBS
- Viability stain (e.g., Propidium Iodide)
- Sterile cell strainer (50 μm pore size)
- Sterile flow cytometry and collection tubes
Procedure:
- Cell Preparation: Plate the mutant cell library a day before sorting to ensure optimal cell health and confluency. On sorting day, harvest adherent cells by aspirating media, washing with PBS, and adding Accutase (1 mL/10-cm dish). Incubate at 37°C for 5-10 minutes until cells detach. Neutralize with complete media. For suspension cells, gently pipette to dissociate clumps [25].
- Washing and Counting: Transfer cell suspension to a conical tube and centrifuge at 300 × g at 4°C for 10 minutes. Aspirate supernatant and resuspend cells in complete media. Count live cells using a hemocytometer and Trypan Blue exclusion.
- Viability Staining: Wash cells twice with ice-cold PBS. Resuspend the final cell pellet in FACS Buffer containing a viability stain (e.g., Propidium Iodide) according to the manufacturer's recommendation. Incubate on ice for 15-30 minutes, protected from light.
- Final Resuspension and Filtration: Centrifuge cells and resuspend in FACS Buffer at a concentration of 1–2 × 10⁷ cells/mL. Gently pipette to disrupt clumps. Pass the cell suspension through a sterile 50 μm cell strainer into a sterile flow cytometry tube to remove any remaining aggregates.
- FACS Sorting: Keep samples on ice and protected from light. Use the unlabeled and single-color controls to set up the instrument and compensate for fluorescence spillover. Implement a sequential gating strategy: (1) FSC-A vs. SSC-A to gate on cells and exclude debris, (2) FSC-H vs. FSC-W to select single cells, (3) viability dye-negative to select live cells, and finally (4) GFP-positive vs. GFP-negative to define the target populations. Sort the desired populations into collection tubes containing an appropriate recovery medium.
- Post-Sort Processing: Return collected cells to sterile culture conditions as soon as possible. It is critical to validate the sort purity by re-analyzing an aliquot of the sorted cells and to confirm the CRISPR editing efficiency via genomic PCR, T7E1 assay, or sequencing.
Alternate Protocol: Sorting GFP+ Cells from Autofluorescent Populations
This protocol is crucial for working with inherently autofluorescent cells, such as Retinal Pigment Epithelium (RPE) cells, which accumulate autofluorescent granules like lipofuscin [26].
Materials: (In addition to Basic Protocol materials)
- Specific antibodies for potential cell surface markers (optional)
Procedure:
- Cell Preparation and Staining: Follow Steps 1–3 of the Basic Protocol to generate a single, viable cell suspension.
- Advanced Gating Strategy: The key differentiator is the use of a more sophisticated gating approach to distinguish true GFP signal from autofluorescence.
- After selecting for single, live cells, create a plot of the GFP signal (typically FITC or Alexa Fluor 488 channel) versus the signal from a fluorescent channel adjacent to GFP (e.g., PE or PE-Texas Red).
- Autofluorescent granules will typically emit signals detectable in both channels, appearing along a diagonal in this plot.
- True GFP-positive cells will be positive for GFP but low for the adjacent channel, forming a distinct population that can be gated accordingly [26].
- Sorting and Validation: Proceed with sorting as in the Basic Protocol. Post-sort validation is especially critical in this context to confirm that the sorted "GFP-positive" population is indeed enriched for the target cells and not for highly autofluorescent untransfected cells.
Troubleshooting and Technical Considerations
- Low Cell Viability Post-Sort: Ensure the FACS buffer contains EDTA and protein (e.g., FBS, BSA), all solutions are ice-cold, and the time from cell harvesting to final collection is minimized. Maintaining sorted cells on ice after collection is also vital [25].
- Poor Sort Purity: Always include control samples (untransfected/unlabeled cells) to accurately set sorting gates. The use of a viability dye is non-negotiable for excluding dead cells, which often exhibit non-specific fluorescence and stick to live cells, causing contamination. Re-analysis of a sorted sample aliquot is the best practice to confirm purity.
- Low Yield/High Cell Loss: FACS is inherently a lower-yield method compared to magnetic sorting (MACS), with reported cell losses around ~70% for FACS versus 7–9% for MACS [28]. If high cell numbers are the priority for downstream applications, consider a pre-enrichment step with MACS where possible, or ensure you begin with a sufficiently large input cell number.
- Sample Transfer Time: For samples being processed off-site, shorter transfer times significantly improve FACS success. One study on multiple myeloma samples showed a 77.1% success rate for transfer times <2 hours, compared to 67.8% for longer times [30].
In CRISPR-based functional genomics, the design of sgRNA libraries and the achievement of sufficient sgRNA coverage are fundamental to screening success. Library design determines the comprehensiveness and specificity of genetic perturbations, while coverage ensures that screening results are statistically robust and reproducible. These factors are particularly crucial in visual screening systems utilizing fluorescent markers like eGFP, where precise editing outcomes must be accurately quantified across large cell populations. Optimal library design and coverage enable researchers to distinguish between different DNA repair outcomes, identify key genetic regulators, and unravel complex biological mechanisms in disease contexts such as cancer [31] [32] [33].
The integration of visual reporters like eGFP provides a powerful tool for rapid assessment of editing efficiency. In these systems, successful homology-directed repair (HDR) can convert eGFP to blue fluorescent protein (BFP), while non-homologous end joining (NHEJ) typically results in loss of fluorescence, creating a dual-readout system that enables high-throughput screening of editing outcomes [14] [32]. This approach allows researchers to simultaneously evaluate both gene knockout and specific gene correction events, providing critical insights for developing genome editing therapies.
Principles of sgRNA Library Design
Library Size and Composition
Effective sgRNA libraries balance comprehensiveness with practical feasibility. Genome-wide libraries systematically target thousands of genes, while focused libraries interrogate specific pathways or gene sets. The number of sgRNAs per gene represents a critical design parameter, with traditional libraries employing 4-10 sgRNAs per gene to ensure effective perturbation [33]. However, recent advances demonstrate that smaller, more optimized libraries can perform equivalently or superior to larger conventional libraries.
Table 1: Comparison of CRISPR Library Designs and Performance
| Library Name | sgRNAs per Gene | Library Size | Performance Notes | Optimal Use Cases |
|---|---|---|---|---|
| Brunello [33] | 4 | Standard | Balanced performance | General genome-wide screening |
| Yusa v3 [33] | 6 | Large | Comprehensive but lower efficacy in some tests | Applications requiring maximum coverage |
| Vienna-single [33] | 3 | Reduced by 50% | Stronger depletion of essential genes than larger libraries | Cost-sensitive studies; limited cell material |
| Vienna-dual [33] | 3 pairs | Reduced by 50% | Strongest performance in essentiality and drug-gene interaction screens | Enhanced knockout efficiency needed |
| MinLib [33] | 2 | Minimal | Potential best performance per guide | Extreme library compression required |
Recent benchmarking studies reveal that libraries with fewer sgRNAs per gene, when selected using principled criteria like Vienna Bioactivity CRISPR (VBC) scores, can outperform larger conventional libraries. The top3-VBC library (3 guides per gene) demonstrated stronger depletion of essential genes than the Yusa v3 6-guide library, highlighting that guide quality supersedes quantity [33]. This library compression enables more cost-effective screens with reduced reagent and sequencing costs, increased throughput, and improved feasibility for applications with limited material such as organoids or in vivo models.
Dual vs. Single Targeting Strategies
Dual-targeting libraries, where two sgRNAs target the same gene, can enhance knockout efficiency through deletion of the inter-sgRNA genomic region. Evidence indicates that dual-targeting guides produce stronger depletion of essential genes and weaker enrichment of non-essential genes compared to single-targeting approaches [33]. However, this strategy may trigger a heightened DNA damage response due to creating twice the number of double-strand breaks, potentially introducing confounding fitness effects in certain screening contexts.
The performance advantage of dual-targeting appears most pronounced when pairing less efficient guides with more efficient ones, effectively compensating for variable guide efficacy. Interestingly, the benefit of dual-targeting was largely absent when using the highly efficient Vienna-single library guides, suggesting that the approach provides maximal benefit when guide efficacy is suboptimal [33]. The distance between gRNA pairs, either in absolute terms or relative to gene length, shows no clear correlation with efficacy, contradicting earlier reports [33].
Calculating and Achieving Sufficient sgRNA Coverage
Coverage Fundamentals and Requirements
Coverage refers to the number of cells representing each sgRNA in a library, determining the statistical power to detect phenotypic effects. The established gold standard for genome-wide knockout screens is 250x coverage—meaning each unique sgRNA is represented in at least 250 cells [34]. This threshold ensures sufficient representation to distinguish true phenotypic effects from stochastic noise.
Coverage requirements directly determine library size and screening scale. For a genome-wide library targeting ~20,000 human genes with 4 sgRNAs per gene, achieving 250x coverage requires delivering sgRNAs to at least 20 million cells (20,000 genes × 4 sgRNAs × 250 cells) [34]. This substantial cell requirement presents significant challenges for in vivo screens or models with limited cell availability.
Table 2: Coverage Requirements and Computational Tools
| Parameter | Standard Requirement | Minimum Viable | Calculation Basis |
|---|---|---|---|
| Coverage per sgRNA | 250x [34] | Variable by screen type [34] | Statistical power to detect phenotype |
| Cells for genome-wide screen (4 sgRNAs/gene) | 20 million [34] | Lower with optimized libraries [33] | (20,000 genes × 4 sgRNAs × 250 cells) |
| Guide efficacy prediction | VBC scores [33] | Rule Set 3 [33] | Correlation with log-fold changes |
| Performance assessment | Chronos algorithm [33] | MAGeCK [33] | Gene fitness estimates across time series |
Strategies for Optimizing Coverage
Innovative approaches can reduce coverage requirements while maintaining screening quality:
Library Compression: Using minimal libraries with 2-3 highly effective sgRNAs per gene dramatically reduces cell requirements. The Vienna library (3 guides/gene) achieves 250x coverage with approximately 15 million cells for genome-wide screening—a 25% reduction compared to standard 4-guide libraries [33].
Pooled Screening Across Organisms: For in vivo applications where target cells are limited, distributing a genome-wide library across multiple animals can achieve sufficient coverage. One approach divides the library into sub-libraries, each delivered to a separate organism [34]. Alternatively, delivering the complete library to multiple animals increases aggregate coverage while introducing inter-organism variability [34].
Retrospective Coverage Analysis: Analysis of T cell screening data suggests that fitness phenotypes may be detectable below the 250x standard, indicating that required coverage is screen-specific [34]. Factors influencing minimum requirements include cell population heterogeneity and phenotypic selection strength.
Protocol: eGFP to BFP Mutation Reporter System for Assessing Editing Outcomes
Experimental Workflow and Principles
The eGFP to BFP mutation reporter system provides a visual method to simultaneously quantify HDR and NHEJ outcomes in CRISPR-edited cells. This system utilizes a lentivirally delivered eGFP construct integrated into the cellular genome. Designed sgRNAs target the eGFP sequence, while HDR templates introduce specific nucleotide substitutions that convert eGFP to BFP. Successful HDR generates blue fluorescent cells, while NHEJ produces non-fluorescent cells, enabling quantitative assessment of editing outcomes via flow cytometry [14] [32].
Step-by-Step Methodology
Generation of eGFP-Positive Cell Lines
Materials:
- HEK293T cells (or other relevant cell lines)
- Lentiviral vectors: pMD2.G, pRSV-Rev, pMDLg/pRRE, pHAGE2-Ef1a-eGFP-IRES-PuroR [32]
- Polyethylenimine (PEI) transfection reagent
- Complete DMEM medium with 10% FBS
- Puromycin selection antibiotic
Procedure:
- Cell Culture Preparation: Thaw and culture HEK293T cells in complete DMEM medium. Maintain cells below 80% confluency with regular passaging every 3-4 days using trypsin-EDTA detachment [32].
- Lentivirus Production: Co-transfect HEK293T cells with the lentiviral packaging plasmids (pMD2.G, pRSV-Rev, pMDLg/pRRE) and the pHAGE2-Ef1a-eGFP-IRES-PuroR transfer plasmid using PEI transfection reagent [32].
- Viral Harvest and Transduction: Collect viral supernatant 48-72 hours post-transfection. Filter through 0.45μm membrane and transduce target cells with viral supernatant supplemented with 8μg/mL polybrene.
- Selection of eGFP-Positive Cells: Begin puromycin selection (2μg/mL) 48 hours post-transduction. Maintain selection for 5-7 days until non-transduced control cells are completely eliminated [32].
- Validation: Confirm eGFP expression using fluorescence microscopy or flow cytometry before proceeding with editing experiments.
CRISPR-Cas9 Transfection and Editing
Materials:
- Purified SpCas9-NLS protein [32]
- sgRNA targeting eGFP: 5'-GCUGAAGCACUGCACGCCGU-3' [32]
- HDR template: 5'-caagctgcccgtgccctggcccaccctcgtgaccaccctgAGCCACggcgtgcagtgcttcagccgctaccccgaccacatgaagc-3' (mutations shown in uppercase) [32]
- Transfection reagent (e.g., ProDeliverIN CRISPR)
Procedure:
- RNP Complex Formation: Combine SpCas9 protein (10μg) and sgRNA (5μg) in opti-MEM medium. Incubate at room temperature for 10-15 minutes to form ribonucleoprotein (RNP) complexes [32].
- HDR Template Preparation: Dilute single-stranded oligodeoxynucleotide (ssODN) HDR template in opti-MEM to appropriate working concentration.
- Cell Transfection: Harvest eGFP-positive cells and resuspend in appropriate transfection medium. Combine RNP complexes and HDR template with cell suspension using transfection reagent according to manufacturer's protocol.
- Post-Transfection Culture: Plate transfected cells in complete medium and incubate at 37°C, 5% CO2 for 72-96 hours to allow expression of editing outcomes [32].
Analysis of Editing Outcomes
Materials:
- Flow cytometry buffer (PBS with 1% BSA)
- 4% paraformaldehyde fixation solution
- Flow cytometer with 488nm (eGFP) and 405nm (BFP) lasers
Procedure:
- Sample Preparation: Harvest cells 72-96 hours post-transfection. Wash with PBS and resuspend in flow cytometry buffer. Optionally fix cells with 4% PFA for 15 minutes if analysis cannot be performed immediately [32].
- Flow Cytometry Analysis: Analyze cells using flow cytometry equipped with appropriate lasers and filters. Detect eGFP fluorescence using 488nm excitation and 530/30nm emission. Detect BFP fluorescence using 405nm excitation and 450/50nm emission [32].
- Data Interpretation: Identify three distinct populations:
- BFP-positive: Successful HDR (template-mediated repair)
- eGFP-positive: Unedited or perfectly repaired cells
- Non-fluorescent: NHEJ-mediated indels causing frameshift mutations
- Efficiency Calculation:
- HDR efficiency = (BFP+ cells / total viable cells) × 100
- NHEJ efficiency = (non-fluorescent cells / total viable cells) × 100
- Total editing efficiency = HDR efficiency + NHEJ efficiency
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for CRISPR Screening with Fluorescent Reporters
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| CRISPR Effectors | SpCas9-NLS [32] | Creates double-strand breaks at target DNA sequences guided by sgRNA |
| Delivery Tools | ProDeliverIN CRISPR [32], PEI (polyethylenimine) [32] | Enables efficient intracellular delivery of CRISPR components |
| Visual Reporters | eGFP, BFP mutation system [14] [32] | Provides rapid, quantitative readout of editing outcomes via fluorescence changes |
| Library Design Tools | VBC scores [33], Rule Set 3 [33] | Predicts sgRNA efficacy for optimal library design |
| Selection Agents | Puromycin [32] | Selects for successfully transduced cells when using lentiviral systems |
| Analytical Tools | FlowLogic, GraphPad Prism [32] | Analyzes flow cytometry data and performs statistical analysis |
| HDR Templates | ssODNs with specific mutations [32] | Serves as repair template for precise genome editing via HDR |
| Cell Lines | HEK293T, HepG2, IMR90 [32] | Provides cellular context for screening; HEK293T commonly used for lentivirus production |
Strategic library design and adequate sgRNA coverage form the foundation of successful CRISPR screening. The movement toward smaller, more optimized libraries—such as the 3-guide Vienna library—demonstrates that guide quality and selection criteria significantly outweigh mere quantity in screening performance. The integration of visual screening systems utilizing eGFP to BFP conversion provides a robust, high-throughput method for quantifying editing outcomes, enabling rapid optimization of editing conditions and formulations. As CRISPR screening evolves, these design principles and experimental approaches will continue to enhance our ability to systematically interrogate gene function and develop novel therapeutic strategies.
In the field of visual screening for CRISPR transformants, the generation of GFP-enriched cell pools represents a powerful strategy for high-throughput functional genomics research. This approach allows researchers to study gene expression, protein localization, and cellular responses to perturbations while preserving parental cell line heterogeneity. A critical technical consideration in these experiments is determining the optimal sequencing depth for accurately characterizing the genetic composition of GFP-enriched pools. Proper sequencing depth ensures comprehensive detection of CRISPR-induced mutations, precise identification of successfully tagged clones, and reliable quantification of sgRNA abundances, all of which are essential for robust experimental outcomes in drug discovery and basic research applications. This application note details the methodologies and sequencing parameters required for effective analysis of GFP-enriched pools within the broader context of CRISPR transformant screening.
Experimental Design for GFP-Enriched Pool Generation
Strategies for Generating Homozygous Fluorescent Reporter Cell Pools
The creation of GFP-enriched cell pools begins with the implementation of precise genome editing techniques. Researchers have developed multiple strategies to engineer homozygous fluorescent reporter knock-in cell pools that circumvent the clonal variability inherent to traditional approaches. These methodologies share the common goal of achieving biallelic editing with precise genome editing, which is particularly crucial when studying genes with low endogenous expression levels or when homogeneous populations are required for downstream applications [4].
Three primary strategies have been optimized for this purpose:
Dual-plasmid electroporation system: This approach employs separate plasmids for donor DNA and doxycycline-inducible sgRNA expression, delivered via electroporation. While effective for targeted integration, this method is associated with higher rates of random integration, which can complicate subsequent analysis [4].
Single-plasmid electroporation system: This simplified system integrates both sgRNA and donor DNA components into a single vector, incorporating a fluorescent protein marker to help eliminate undesired random integration events. This system reduces complexity while maintaining editing efficiency [4].
Integrase-deficient lentivirus vector (IDLV) system: This delivery method couples a single-plasmid construct with an IDLV packaging system, offering flexibility between electroporation and lentivirus transduction. Notably, the IDLV system significantly minimizes random integration while maintaining high editing efficiency, making it particularly valuable for hard-to-transfect cell types [4].
Donor DNA Design Optimization
A critical aspect of generating high-quality GFP-enriched pools is the optimization of donor DNA design to reduce false-positive cells associated with random integration. The recommended strategy includes:
- Removal of the start codon from the fluorescent reporter sequence to prevent independent translation initiation
- Incorporation of a self-cleaving T2A peptide system to ensure coordinated expression of the endogenous gene and the fluorescent reporter
- Utilization of fluorescence-activated cell sorting (FACS) to efficiently identify and isolate desired homozygous fluorescent knock-in clones, establishing stable cell pools that preserve parental cell line heterogeneity [4]
Multicolour Tagging Approaches
For more complex screening applications, pooled multicolour tagging strategies enable the simultaneous monitoring of multiple proteins or cellular compartments. This approach involves:
- Sequential rounds of intron tagging using orthogonal sgRNA libraries targeting different intron frames
- Implementation of complementary fluorescent tags (e.g., GFP, mScarlet) to create visual barcodes for different proteins
- Addition of structural markers such as membrane-targeted (mAmetrine-CAAX) and nuclear markers (NLS-miRFP670-miRFP670nano) to facilitate cell segmentation during image analysis
- Computer vision and machine learning to identify clones based on localization patterns and expression levels of tagged proteins, enabling simultaneous live-cell monitoring of large protein sets [35]
Sequencing Parameters and Depth Requirements
Key Sequencing Considerations for GFP-Enriched Pools
The determination of optimal sequencing depth for GFP-enriched pools depends on several experimental factors, including pool complexity, tagging efficiency, and the specific research questions being addressed. While the search results do not provide explicit numerical depth recommendations, they highlight critical technical parameters that inform sequencing strategy design.
Table 1: Key Sequencing Parameters for CRISPR-Modified GFP-Enriched Pools
| Parameter | Specification | Application Context |
|---|---|---|
| Amplicon Size Range | 200-280 base pairs | Optimal for CRISPR amplicon sequencing [36] |
| Library Complexity | Varies by sgRNA library size (e.g., 90,657 sgRNAs in genome-wide library) | Dependent on experimental scale [35] |
| Tagging Efficiency | Typically 10-20% of positive control sgRNAs in pooled format | Affects required depth for rare event detection [35] |
| Variant Detection | Ultra-deep sequencing for sensitive indel detection | Essential for characterizing editing efficiency [36] |
| Multiplexing Capacity | Sample multiplexing using validated indices | Enables high-throughput processing [36] |
Amplicon Sequencing for Mutation Verification
For verification of CRISPR-induced mutations in GFP-enriched pools, amplicon sequencing provides a sensitive and cost-effective approach:
- Target-specific amplicon design focusing on 200-280 bp regions surrounding the CRISPR target site
- Elimination of cloning steps through direct amplification from pooled cellular material
- Ultra-deep sequencing to detect even low-frequency mutations within heterogeneous pools
- Specialized variant calling algorithms optimized for CRISPR-induced mutation patterns [36]
The high sensitivity of this approach enables researchers to quantify editing efficiencies and identify potential off-target effects while processing multiple samples in parallel through index-based multiplexing.
Research Reagent Solutions
Table 2: Essential Research Reagents for GFP-Enriched Pool Generation and Sequencing
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| CRISPR Delivery Systems | Dual-plasmid system; Single-plasmid system; IDLV system [4] | Delivery of editing components with varying random integration rates |
| Fluorescent Reporters | EGFP; miRFP670; mScarlet; BFP [4] [35] | Visual tagging of endogenous proteins for tracking and sorting |
| sgRNA Libraries | Genome-wide intron-targeting (90,657 sgRNAs); Focused cancer libraries [35] | Targeted disruption or tagging of specific genomic loci |
| Donor Templates | Minicircle donor DNA; pw35P2AGal4; pBPGAL4.2::p65Uw [35] [37] | Homology-directed repair templates for precise editing |
| Selection Markers | P2A peptide system; mini-white cassette; P3-DsRed [37] | Identification and selection of successfully edited cells |
| Sequencing Reagents | Validated indices; Amplicon sequencing primers [36] | Multiplexed deep sequencing of edited genomic regions |
Experimental Workflow and Protocol
Comprehensive Protocol for GFP-Enriched Pool Generation and Sequencing
Stage 1: Pooled CRISPR Screening and GFP Enrichment
- sgRNA Library Transduction: Transduce target cells with your selected sgRNA library using appropriate delivery methods. For hard-to-transfect cells, consider the IDLV system to minimize random integration [4].
- CRISPR Complex Delivery: Co-transfect with Cas9-expressing plasmid and minicircle donor DNA containing the GFP cassette. Minicircle DNA improves tagging efficiency approximately twofold compared to plasmid donors and reduces backbone integration [35].
- Fluorescence-Based Sorting: After sufficient expression time (typically 7-14 days), use FACS to isolate GFP-positive cells. Gate stringently to enrich for cells with successful knock-in events [4].
- Expansion of Enriched Pool: Culture sorted cells to expand the GFP-enriched pool while maintaining adequate representation of all integrated variants.
Stage 2: Sequencing Preparation and Analysis
- Genomic DNA Extraction: Harvest cells from the expanded GFP-enriched pool and extract genomic DNA using standard methods.
- Target-Specific PCR Amplification: Design primers flanking the integrated GFP sequence and CRISPR target site. Amplify regions of 200-280 bp for optimal sequencing results [36].
- Library Preparation and Multiplexing: Incorporate validated indices during library preparation to enable sample multiplexing. This allows parallel processing of multiple experimental conditions or time points [36].
- High-Throughput Sequencing: Sequence the prepared libraries using an appropriate sequencing platform. The required depth depends on pool complexity but should be sufficient to detect even low-abundance integration events.
- Bioinformatic Analysis: Process sequencing data to determine sgRNA abundance distribution, verify precise integration events, and quantify editing efficiencies across the pool.
Workflow Visualization
Diagram 1: GFP-Enriched Pool Sequencing Workflow
Technical Considerations and Optimization
Factors Influencing Sequencing Depth Requirements
Several experimental factors directly impact the determination of optimal sequencing depth for GFP-enriched pools:
- Pool Complexity: The number of distinct sgRNAs or integration events in the pool significantly influences required sequencing depth. Genome-wide screens with >90,000 sgRNAs require greater depth than focused libraries targeting specific gene sets [35].
- Tagging Efficiency Variation: Different sgRNAs exhibit substantial variation in tagging efficiency (up to 18.7% for high-efficiency guides vs. much lower rates for others). This variability necessitates sufficient depth to detect less efficient integration events [35].
- Multimodal Phenotyping Requirements: Integrated approaches that combine sequencing with imaging, such as Perturb-Multimodal, may require adjusted sequencing parameters to correlate genetic perturbations with spatial and morphological phenotypes [38].
Quality Control Metrics
Implement rigorous QC measures throughout the experimental process:
- Verify sgRNA Representation: After FACS enrichment, assess whether the distribution of sgRNAs in the GFP-positive population reflects expected patterns based on tagging efficiency.
- Monitor Contamination: Include control sgRNAs that should not generate functional GFP fusions to assess background signal and random integration.
- Validate Integration Precision: Use sequencing data to confirm precise integration at target loci and quantify the rate of spurious integration events.
The determination of optimal sequencing depth for GFP-enriched pools represents a critical methodological consideration in visual CRISPR screening approaches. While specific depth requirements vary based on experimental parameters, the fundamental goal remains achieving sufficient coverage to comprehensively characterize the genetic diversity within enriched pools. By implementing the strategies outlined in this application note—including optimized donor design, appropriate delivery systems, and sensitive amplicon sequencing—researchers can reliably generate and sequence GFP-enriched pools for diverse functional genomics applications. These methodologies support the growing emphasis on pooled screening approaches that preserve cellular heterogeneity while enabling high-throughput analysis of gene function and protein dynamics in relevant biological contexts.
The advent of CRISPR-Cas technology has revolutionized functional genomics by enabling precise, scalable, and programmable genome engineering. A particularly powerful application lies in genome-wide screens and multi-gene knockout strategies, which allow researchers to systematically interrogate gene function at an unprecedented scale. Unlike single-gene editing approaches, multiplexed CRISPR-Cas systems facilitate the simultaneous targeting of multiple genomic loci using guide RNA (gRNA) arrays, enabling the functional analysis of complex genetic networks, synthetic lethal interactions, and non-coding elements that would otherwise remain elusive [39].
These advanced screening methodologies are increasingly integrated with visual reporter systems, such as green fluorescent protein (GFP), to rapidly quantify editing outcomes and efficiency. The convergence of multiplexed gRNA delivery with fluorescent-based readouts provides a robust platform for identifying genetic determinants of disease and potential therapeutic targets, particularly in drug development pipelines where understanding functional gene interactions is paramount [14] [40].
Multiplexed Screening Strategies and Experimental Workflows
Dual-Targeting Strategies for Gene Knockout
A fundamental advance in CRISPR screening has been the development of dual-targeting strategies to ensure complete gene disruption. While single-guide RNA (sgRNA) approaches can generate small insertions or deletions (indels) via error-prone non-homologous end joining (NHEJ), they may not always result in functional knockouts. Dual-gRNA systems address this limitation by introducing two simultaneous double-strand breaks within a single gene, producing a large genomic deletion that unequivocally disrupts the coding sequence [39].
The workflow for a typical dual-gRNA knockout experiment involves:
- Designing two gRNAs targeting distinct exons of the target gene
- Cloning both gRNAs into a single delivery vector
- Transducing the target cell population
- Validating knockout efficiency via PCR and sequencing
- Phenotypic screening to assess functional consequences
This approach has proven particularly valuable for studying long non-coding RNAs (lncRNAs) and other non-coding elements, where complete excision of the genomic locus is often necessary to elucidate function [39].
Genome-Wide Screens with Combinatorial Knockout
For comprehensive functional genomics, researchers have developed genome-scale combinatorial knockout libraries that systematically target gene pairs. The CRISPR-based double-knockout (CDKO) library represents a sophisticated implementation of this approach, employing carefully engineered lentiviral vectors that express two gRNAs from different polymerase III promoters (e.g., human U6 and mouse U6) to minimize recombination events [39].
These libraries enable the systematic identification of synthetic lethal interactions - where simultaneous disruption of two genes is lethal while individual knockouts are viable - with profound implications for cancer therapy development. In one notable application, a CDKO library screening 490,000 gRNA pairs in K562 cells successfully identified synthetic lethal interactions with specific therapeutic compounds [39].
Workflow: Genome-Wide Combinatorial Screen
The following diagram illustrates the key steps in a genome-wide combinatorial CRISPR screen:
Quantitative Data from Multiplexed Editing Studies
Editing Efficiencies Across Experimental Systems
Multiplexed CRISPR approaches have demonstrated high efficiency across diverse biological systems, from microbial organisms to mammalian cells. The table below summarizes key performance metrics from recent studies:
Table 1: Efficiency Metrics of Multiplexed CRISPR-Cas Editing Systems
| Organism/System | Editing Type | Targets | Efficiency | Application | Reference |
|---|---|---|---|---|---|
| Pichia pastoris | Dual-gene knockout | 2 genes | 60-100% | Metabolic engineering | [41] |
| Tobacco plants | SMG cassette excision | 4 gRNAs | ~10% | Marker-free transgenic plants | [42] |
| Human cell lines | Dual-gene knockout | 2 genes | Varies by locus | Functional genomics | [39] |
| GFP-on mouse model | Base editing correction | Single nucleotide | ~50% (in fibroblasts) | In vivo editing validation | [40] |
| Human K562 cells | Combinatorial screening | 490,000 gRNA pairs | Identification of synthetic lethals | Drug target discovery | [39] |
Comparison of Multi-gRNA Expression Systems
The efficiency of multiplexed editing critically depends on the strategy for expressing multiple gRNAs from a single vector. Different approaches have been developed and optimized for specific applications:
Table 2: Comparison of Multi-guide RNA Expression Systems
| Expression System | Mechanism | Maximum Guides Demonstrated | Advantages | Limitations |
|---|---|---|---|---|
| tRNA-gRNA array | Endogenous tRNA processing | 10+ | High efficiency, modular | Potential context effects |
| HgH structure | Ribozyme-mediated processing | 4+ | Consistent processing | More complex cloning |
| Dual polymerase III promoters | Separate transcriptional units | 2 | Prevents recombination | Limited to 2 guides without modification |
| Csy4-based system | Protein-mediated cleavage | 10+ | Highly specific | Requires Csy4 co-expression |
Experimental Protocols
Protocol: Multiplexed CRISPR Screening with Fluorescent Reporters
This protocol outlines the steps for conducting a multiplexed CRISPR screen using GFP-based visual screening of editing outcomes, adapted from established methodologies [14] [40] [43].
Preparation of gRNA Expression Vectors
- Design gRNAs targeting genes of interest using validated computational tools (e.g., CRISPick, CHOPCHOP)
- Clone gRNA sequences into appropriate expression vectors using Golden Gate assembly or similar modular cloning strategies
- For dual-gRNA vectors, use heterologous promoters (e.g., human U6 and mouse U6) to prevent homologous recombination
- Incorporate fluorescent reporter genes (e.g., GFP, BFP, DsRED) for tracking transduction efficiency and editing outcomes
Library Delivery and Cell Selection
- Transduce target cells at low multiplicity of infection (MOI < 0.3) to ensure most cells receive a single vector
- Apply selection (e.g., puromycin, blasticidin) 24-48 hours post-transduction to eliminate untransduced cells
- Monitor fluorescence to confirm reporter expression and transduction efficiency
- Harvest cells at appropriate time points for genomic DNA extraction and phenotypic analysis
Screening and Hit Validation
- Extract genomic DNA using standardized protocols suitable for next-generation sequencing
- Amplify gRNA regions with primers containing Illumina adapter sequences
- Sequence libraries on an appropriate Illumina platform to determine gRNA abundance
- Identify significantly enriched or depleted gRNAs using specialized algorithms (e.g., MAGeCK, BAGEL)
- Validate hits using individual gRNAs in secondary screens
Protocol: Visual Assessment of Editing Efficiency via GFP Conversion
The following workflow enables rapid screening of CRISPR-Cas9 editing outcomes by mutating enhanced GFP to blue or non-fluorescent phenotypes [14]:
Establishing Reporter Cell Lines
- Generate eGFP-expressing cell lines through lentiviral transduction or stable transfection
- Validate uniform eGFP expression using flow cytometry and fluorescence microscopy
- Clone gRNAs targeting critical residues in the eGFP coding sequence that, when mutated, alter fluorescence properties
Editing and Analysis
- Deliver CRISPR-Cas9 components and gRNAs to eGFP-expressing cells via transfection or transduction
- Monitor fluorescence changes 72-96 hours post-delivery using flow cytometry or high-content imaging
- Sort cells based on altered fluorescence (eGFP-negative or BFP-positive) for downstream analysis
- Sequence target loci to correlate fluorescence changes with specific mutation types
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of multiplexed CRISPR screens requires carefully selected reagents and tools. The following table outlines key components:
Table 3: Essential Research Reagents for Multiplexed CRISPR Screening
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Cas9 variants | Wild-type Cas9, Nickase Cas9, Base editors | DNA cleavage or modification | Nickase Cas9 reduces off-target effects in dual-gRNA approaches [39] |
| gRNA expression systems | U6 promoter, tRNA-gRNA arrays, HgH structure | Express multiple guide RNAs | HgH structure achieved 95.8% single-gene knockout efficiency in P. pastoris [41] |
| Delivery vehicles | Lentivirus, AAV, Electroporation | Introduce editing components | AAV9 enables in vivo delivery in mouse models [40] |
| Fluorescent reporters | eGFP, BFP, DsRED | Visual assessment of editing | eGFP to BFP conversion enables rapid editing assessment [14] |
| Selection markers | Puromycin N-acetyltransferase, Aminoglycoside phosphotransferase | Enumerate edited cells | Can be excised using multiplex CRISPR after selection [42] |
| Screening libraries | CDKO library, Human whole-genome libraries | Genome-scale screening | CDKO library contains 490,000 gRNA pairs [39] |
Workflow: Dual-Guide RNA Mediated Gene Knockout
The molecular workflow for dual-guide RNA mediated gene knockout illustrates the process from cellular delivery to phenotypic validation:
Applications in Drug Development and Therapeutic Discovery
Multiplexed CRISPR screening technologies have transformative potential in pharmaceutical research and development. By enabling systematic mapping of genetic interactions and synthetic lethal relationships, these approaches facilitate:
- Target Identification: Genome-wide screens can reveal novel therapeutic targets across diverse disease areas, particularly in oncology where genetic dependencies vary between cancer types
- Combination Therapy Development: Identification of synthetic lethal interactions informs rational design of combination therapies that maximize efficacy while minimizing resistance
- Biomarker Discovery: Screens can identify genetic modifiers of drug response, enabling patient stratification strategies and personalized medicine approaches
- Toxicology Assessment: Understanding genetic networks helps predict potential adverse effects and off-target activities of candidate compounds
The integration of fluorescent reporter systems with multiplexed editing further streamlines drug discovery pipelines by enabling rapid, high-throughput assessment of editing efficiency and functional outcomes without requiring lengthy sequencing validation at initial screening stages [14] [40]. This is particularly valuable in large-scale compound screens where rapid readouts are essential for prioritizing hits for further investigation.
The pursuit of robust, high-throughput methods to identify key developmental regulators is a central goal in modern stem cell and developmental biology. The integration of fluorescent reporter systems with CRISPR-based functional genomics has emerged as a powerful strategy to address this challenge. This case study details a specific implementation of this approach, focusing on a genome-wide CRISPR knockout screen that utilized a PAX6::H2B-GFP reporter line in human embryonic stem cells (hESCs) to identify novel regulators of developmental timing [44]. The PAX6 transcription factor serves as a critical marker for neuroectoderm differentiation, making it an ideal sentinel for tracking the pace of this fundamental developmental process.
This research exemplifies the broader thesis that visual screening of CRISPR transformants with GFP markers provides an unparalleled window into dynamic biological processes. By enabling real-time tracking of gene expression in living cells, this methodology transforms our ability to deconstruct complex developmental timelines and identify their molecular controllers with high precision and scalability.
Experimental Design & Workflow
Core Experimental Strategy
The experimental design employed a comprehensive approach to identify genes regulating the speed of human neuroectoderm differentiation. Researchers engineered a H9 hESC line containing two critical genetic modifications: (1) a PAX6::H2B-GFP reporter construct that accurately reflects endogenous PAX6 expression dynamics, and (2) a doxycycline-inducible Cas9 (iCas9) system integrated into the AAVS1 safe harbor locus for precise temporal control of gene editing [44]. This dual system enabled the researchers to perturb gene function across the entire genome while simultaneously monitoring the differentiation status of living cells through GFP expression.
The screening strategy leveraged the well-characterized dual SMAD inhibition protocol for directed differentiation of hESCs into neuroectoderm. This protocol yields nearly 100% conversion efficiency with a predictable temporal progression of PAX6 expression, making it ideal for quantifying acceleration or deceleration of developmental pace [44].
Detailed Screening Protocol
Step 1: Library Transduction and Mutagenesis
- Culture PAX6::H2B-GFP iCas9 hESCs under standard pluripotency-maintaining conditions.
- Transduce cells at an appropriate multiplicity of infection (MOI ~0.3-0.4) with the Brunello whole-genome CRISPR knockout lentiviral library (approximately 77,441 gRNAs targeting 19,114 genes) [44].
- Select successfully transduced cells with puromycin (concentration: tailored to cell line sensitivity) for 7-10 days.
- Induce Cas9 expression by adding doxycycline (typical concentration: 1-2 μg/mL) for 2 days prior to differentiation initiation.
Step 2: Directed Differentiation and Timing Analysis
- Initiate neuroectoderm differentiation using dual SMAD inhibition protocol with defined media conditions [44] [45].
- Monitor PAX6 expression kinetics via H2B-GFP fluorescence daily using live-cell imaging.
- At precisely defined timepoints (72h and 84h after differentiation induction), when PAX6 expression begins its rapid increase, harvest cells for sorting [44].
Step 3: Fluorescence-Activated Cell Sorting (FACS) and Hit Identification
- Dissociate differentiated cells into single-cell suspension using enzyme-free dissociation buffer.
- Sort cells into PAX6-GFP^high and PAX6-GFP^low populations using FACS with appropriate gating controls.
- Isolate genomic DNA from each sorted population and amplify integrated gRNA sequences via PCR.
- Perform next-generation sequencing and bioinformatic analysis to identify gRNAs enriched in PAX6-GFP^high population using MAGeCK or similar tools [44].
- Validate primary screen hits through individual gRNA infection and differentiation assays.
Table 1: Key Experimental Parameters for Genome-wide Screening
| Parameter | Specification | Purpose/Rationale |
|---|---|---|
| Reporter Line | H9 PAX6::H2B-GFP iCas9 | Tracks neuroectoderm differentiation in live cells |
| CRISPR Library | Brunello whole-genome (~19k genes) | Comprehensive gene coverage with high on-target efficiency |
| Sorting Timepoints | 72h, 84h post-differentiation | Captures initial PAX6 expression increase for acceleration detection |
| Sorted Populations | PAX6-GFP^high vs. PAX6-GFP^low | Identifies mutations causing precocious differentiation |
| Validation Approach | Individual gRNAs & pharmacological inhibition | Confirms screen hits and explores therapeutic potential |
Diagram 1: Visual screening workflow for identifying developmental timing regulators.
Key Findings & Data Analysis
Primary Screen Results and Hit Validation
The genome-wide screen identified 27 high-confidence hits (Z-score > 1, P < 0.05) whose knockout accelerated PAX6 expression during neuroectoderm differentiation [44]. Gene-set enrichment analysis of the screening results revealed significant overrepresentation of genes involved in chromatin remodeling complexes (including ATAC, SET1C, npBAF, and MLL complexes) and mitochondrial metabolic pathways (particularly the tricarboxylic acid cycle) [44].
Secondary validation using individual gRNAs confirmed MEN1 (encoding Menin) and SUZ12 (a component of Polycomb Repressive Complex 2) as the top hits, showing the most substantial fold-increase in PAX6 expression compared to non-targeting controls [44]. Complementary pharmacological validation using small-molecule inhibitors targeting the identified pathways further reinforced these findings:
- Inhibition of PRC2 complex enhanced PAX6 expression
- Disruption of Menin-MLL interaction accelerated neural differentiation
- Combined treatment produced additive effects on acceleration [44]
Table 2: Key Screening Hits and Validation Results
| Gene/Pathway | Function | Fold Change in PAX6 | Mechanistic Insight |
|---|---|---|---|
| MEN1 (Menin) | Scaffold protein in Menin-MLL complex regulating H3K4me3 | High (precise fold-change not specified) | Loss accelerates differentiation; opposes PRC2 function |
| SUZ12 | Essential component of PRC2 regulating H3K27me3 | High (precise fold-change not specified) | Loss accelerates differentiation; balances bivalent domains |
| PRC1/2 Inhibitors | Pharmacological disruption of Polycomb complexes | Significant increase vs. DMSO | Confirms genetic screen results |
| TCA Cycle Genes | Mitochondrial metabolism | Enriched in screen | Links metabolism to developmental timing |
Mechanistic Insights: Epigenetic Regulation of Developmental Pace
Principal Component Analysis of transcriptomic data from wild-type versus mutant differentiations revealed that SUZ12 and MEN1 knockout cells followed an accelerated but parallel trajectory compared to controls, reaching later differentiation stages sooner without dramatic pathway deviation [44].
The molecular mechanism centers on the regulation of bivalent chromatin domains at key developmental gene promoters. These domains harbor both activating (H3K4me3) and repressive (H3K27me3) histone modifications, maintaining genes in a transcriptionally poised state. Menin (through the Menin-MLL complex) promotes H3K4me3, while SUZ12 (through PRC2) catalyzes H3K27me3 [44]. Loss of either factor disrupts this balance, priming developmental genes for faster activation upon differentiation cues.
This mechanism extends beyond neuroectoderm specification. The acceleration effect was consistently observed across definitive endoderm, cardiomyocyte, and neuronal differentiation paradigms, indicating a general role for chromatin bivalency in controlling developmental timing across germ layers and stages [44].
Diagram 2: Molecular mechanism of developmental acceleration via chromatin regulation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for CRISPR/GFP Screening Platforms
| Reagent/Tool | Specification | Application & Function |
|---|---|---|
| PAX6::H2B-GFP Reporter | H2B-tagged GFP for nuclear localization [44] | Live imaging of neuroectoderm differentiation; FACS sorting |
| Inducible Cas9 System | AAVS1-integrated, doxycycline-controlled [44] | Temporal control of mutagenesis; improves viability |
| Brunello Library | Genome-wide (4 gRNAs/gene); high on-target efficiency [44] | Comprehensive gene knockout screening |
| Dual SMAD Inhibition | LDN193189 + SB431542 in defined media [44] [45] | Efficient, synchronized neural differentiation |
| FACS Setup | High-speed sorter with viability detection | Separation of GFP-high and GFP-low populations |
| MAGeCK Algorithm | Computational analysis tool | Identifies significantly enriched gRNAs from NGS data |
Detailed Protocols
PAX6::H2B-GFP Reporter Line Generation and Validation
Principles: The PAX6::H2B-GFP reporter utilizes a histone H2B fusion for nuclear-localized fluorescence, enabling precise quantification of PAX6 expression dynamics and facilitating FACS-based separation of differentiation stages [44] [46].
Step-by-Step Protocol:
- Design targeting construct: Clone H2B-GFP sequence followed by a synthetic polyA signal into a vector containing homology arms targeting the 3' end of the PAX6 coding region, preserving endogenous regulatory elements [46].
- Electroporation: Deliver targeting construct and Cas9 expression vector into H9 hESCs using manufacturer-recommended electroporation parameters.
- Selection and screening: Apply appropriate antibiotic selection (e.g., puromycin 0.5 μg/mL) for 7-10 days. Pick individual colonies and expand for genomic DNA extraction.
- Validate integration: Perform junction PCR using primers external to the homology arms and internal to the GFP sequence. Confirm proper integration without random insertion.
- Functional validation: Differentiate validated clones using dual SMAD inhibition and assess GFP expression dynamics via live imaging and flow cytometry. Compare to endogenous PAX6 expression via immunostaining to confirm reporter fidelity [44].
Genome-wide CRISPR Screen Execution
Principles: This protocol enables systematic identification of genes regulating developmental timing through enrichment analysis of gRNAs in precociously differentiated populations [44].
Step-by-Step Protocol:
- Library amplification and titration: Amplify Brunello library following manufacturer's instructions. Titrate lentiviral particles on target hPSCs to determine MOI achieving 30-40% infection efficiency.
- Screen scale calculation: Ensure >500 cells per gRNA for adequate representation, requiring minimum 4×10⁷ cells for the Brunello library.
- Lentiviral transduction: Incubate cells with lentiviral particles in the presence of polybrene (8 μg/mL) for 24 hours. Replace with fresh medium containing puromycin for selection.
- Cas9 induction and differentiation: Add doxycycline (1-2 μg/mL) 48 hours prior to differentiation induction. Initiate neural differentiation using dual SMAD inhibition protocol.
- Timepoint selection and sorting: At 72h and 84h post-differentiation, dissociate cells and sort PAX6-GFP^high and PAX6-GFP^low populations using FACS. Collect ≥1×10⁷ cells per population for gRNA representation analysis.
- Genomic DNA extraction and sequencing: Extract gDNA using silica column-based methods. Amplify integrated gRNA sequences via PCR with barcoded primers for multiplexing. Sequence on Illumina platform to obtain >500 reads per gRNA.
- Bioinformatic analysis: Process raw sequencing data through MAGeCK pipeline (v0.5.9) to normalize counts and identify significantly enriched gRNAs in PAX6-GFP^high population using robust rank aggregation algorithm [44].
Pharmacological Validation of Screen Hits
Principles: Small molecule inhibitors provide orthogonal validation of genetic screen hits and potential therapeutic applications [44].
Step-by-Step Protocol:
- Inhibitor preparation: Reconstitute small molecules in appropriate solvents per manufacturer specifications. Prepare aliquots to avoid freeze-thaw cycles.
- Dose optimization: Titrate inhibitors (e.g., MI-503 for Menin-MLL; EZH2 inhibitors for PRC2) to determine optimal concentrations that maximize acceleration without inducing cytotoxicity.
- Differentiation with inhibition: Add inhibitors at differentiation initiation. Include DMSO-only controls and non-targeting gRNA controls.
- Quantitative analysis: Assess PAX6 expression at day 4 via flow cytometry. Analyze ≥10,000 events per condition. Calculate fold-change relative to control.
- Combination studies: Test pairwise inhibitor combinations to identify synergistic effects on differentiation acceleration. Use Chou-Talalay method to quantify synergy [44].
Solving Common Challenges in GFP-Based CRISPR Screening
Within visual screening of CRISPR transformants using Green Fluorescent Protein (GFP) markers, a significant artifact can compromise experimental integrity: unexpected GFP expression in promoterless systems. This phenomenon, where GFP expression occurs without a canonical upstream promoter, can lead to false positives and misinterpretation in CRISPR screening data [7]. This Application Note details the origins of this artifact and provides validated protocols to detect, quantify, and mitigate its impact, ensuring the reliability of your research outcomes.
The core of the problem lies in the assumption that GFP will only be expressed when placed under the control of a functional promoter. However, evidence confirms that the enhanced GFP (EGFP) gene can be expressed in mammalian cells even in the apparent absence of a promoter sequence [7]. This aberrant expression can manifest with lower levels and a delayed kinetic profile compared to promoter-driven expression, but it is sufficient to confound high-sensitivity assays commonly used in CRISPR screening [7] [32]. Recognizing and controlling for this artifact is therefore critical for any research involving GFP-based reporter systems, particularly in the development and assessment of genome editing therapies.
Understanding the Artifact and Its Experimental Evidence
The initial evidence for promoterless GFP expression emerged from studies designing lentiviral transfer vectors intended to express GFP only in transduced cells, not in packaging cells [7]. After transfecting the HEK293T packaging cell line, researchers observed unexpected GFP expression. Through a series of controlled experiments, they systematically ruled out potential causes such as auto-transduction, retrotransposon activity, plasmid contamination, and the presence of a cryptic promoter within the vector backbone [7].
The most compelling evidence came from minimizing the transfected DNA fragment to a region containing only "from the start of the GFP gene to 5'LTR R." The GFP gene was expressed again from this minimized fragment, leading to the conclusion that the EGFP coding sequence itself does not require a promoter for expression in this context [7]. The characteristics of this artifact—expression lag and reduced intensity—mean it is easily overlooked or misinterpreted as low-level promoter activity or background noise.
Key Characteristics of Promoterless GFP Expression
The following table summarizes the core findings from the investigation into promoterless GFP expression:
Table 1: Summary of Experimental Evidence for Promoterless GFP Expression
| Experimental Aspect | Observation in Promoterless System | Implication for CRISPR Screening |
|---|---|---|
| Expression Kinetics | Shows a lag and reaches lower levels compared to promoter-driven GFP [7] | Can be mistaken for weak positive or partial gene editing |
| Percentage of Expressing Cells | Reduced percentage of cells show GFP expression [7] | May appear as a heterogeneous cell population, complicating analysis |
| Dependence on Genomic Context | Expression occurred from minimized linear fragments and plasmid backbones [7] | Not dependent on specific vector integration, a risk in various delivery methods |
| Potential Cause | Suggested that the EGFP coding sequence itself may not need a promoter [7] | The artifact is inherent to the GFP reporter itself, not a specific construct design |
Detection and Quantification Methodologies
Protocol: Detecting Promoterless GFP Expression
This protocol is adapted from the methods used to initially characterize the artifact [7] and is a critical first step in validating any GFP-based CRISPR reporter system.
1. Principle: To empirically test if a newly constructed GFP vector or a CRISPR-generated reporter cell line exhibits promoterless expression by transfecting the promoterless construct into a relevant cell line and monitoring for fluorescence.
2. Materials:
- Cells: HEK293T cell line (or your cell line of interest) [7] [32]
- Vectors:
- Experimental: Purified plasmid containing the promoterless GFP cassette.
- Positive Control: Plasmid with GFP under a strong constitutive promoter (e.g., CMV, EF1α) [32].
- Negative Control: "Mock" transfection (no DNA).
- Equipment: Fluorescence microscope, flow cytometer, cell culture incubator.
3. Procedure:
- Day 1: Seed HEK293T cells in a 12-well plate to reach 60-80% confluency at the time of transfection.
- Day 2: Transfert the cells with the promoterless GFP plasmid, the positive control, and the negative control using your standard transfection reagent (e.g., Polyfect) [7].
- Days 3-5: Monitor cells daily for GFP expression using fluorescence microscopy.
- Day 5: Harvest cells and analyze GFP fluorescence using flow cytometry. Compare the fluorescence intensity and the percentage of GFP-positive cells in the experimental group to the positive and negative controls.
4. Analysis and Interpretation:
- The presence of a GFP-positive population in the promoterless transfection group, distinct from the negative control, indicates aberrant expression.
- As reported, the fluorescence intensity will likely be lower, and the percentage of positive cells will be reduced compared to the positive control [7].
Protocol: Absolute Quantification of GFP with FPCountR
To move beyond relative fluorescence units and enable cross-comparisons between instruments and laboratories, absolute quantification of GFP molecules is recommended. The FPCountR method provides a generalizable approach for this [47].
1. Principle: The method uses purified GFP protein as a calibrant to establish a standard curve, converting arbitrary fluorescence units from plate readers into absolute units of molecules of GFP per cell [47].
2. Materials:
- FPCountR Protocol and R Package: Available as open-access tools [47].
- Purified GFP Calibrant: His-tagged GFP expressed from a standardized, inducible vector and purified via His-tag affinity purification [47].
- Protein Quantification Assay: Bicinchoninic acid (BCA) assay or the described 'ECmax' absorbance-based assay [47].
- Equipment: Microplate reader, SDS-PAGE equipment, sonicator.
3. Procedure:
- Produce GFP Calibrant: Express and purify His-tagged GFP from E. coli. Verify purity via SDS-PAGE and confirm fluorescence via excitation/emission scanning [47].
- Determine Protein Concentration: Use the BCA assay (with buffer exchange to avoid interference) or the more robust 'ECmax' assay to determine the precise concentration of the purified GFP [47].
- Generate Standard Curve: Perform a fluorescence assay on a dilution series of the GFP calibrant with known molecular concentration using your calibrated plate reader.
- Analyze Data: Use the FPCountR package to fit the standard curve and obtain a conversion factor (RFU per GFP molecule).
4. Analysis and Interpretation:
- This calibration allows you to convert fluorescence readings from your experimental cells (e.g., CRISPR-edited cells showing unexpected fluorescence) into an absolute number of GFP molecules per cell [47].
- This quantitative data is crucial for determining if the level of aberrant expression is significant enough to impact your specific assay's readout.
Diagram 1: Workflow for detecting and quantifying promoterless GFP expression artifacts.
Mitigation Strategies and CRISPR-Specific Protocols
Strategies to Minimize Artifact Impact
Once detected, the following strategies can help mitigate the risk posed by promoterless GFP expression:
- Incorporate Stringent Controls: Always include promoterless GFP vectors and non-transfected cells as negative controls in every experiment. Set fluorescence thresholds based on these controls to filter out background signal [7].
- Employ Multi-Color Reporter Systems: Instead of relying solely on GFP, use a system that requires a specific mutation for a phenotypic switch. A robust protocol exists where editing an eGFP sequence to convert it to a Blue Fluorescent Protein (BFP) via HDR allows differentiation between non-edited (GFP+), knock-out (non-fluorescent), and successfully edited (BFP+) cells [14] [32].
- Utilize Marker-Free CRISPR Editing: To avoid interference from antibiotic resistance markers and their promoters, use marker-free CRISPR/Cas9 protocols. These systems, which employ positive-negative selection (e.g., using hdhfr-yfcu cassette), allow for the generation of clean transgenic parasites or cell lines without integrated selection markers, reducing the risk of confounding expression [48].
- Validate with Orthogonal Methods: Do not rely on GFP fluorescence as the sole readout. Correlate findings with other methods, such as PCR-based genotyping or Western blotting, to confirm the presence of the intended genetic alteration [48].
Protocol: A CRISPR-Cas9 Fluorescence Conversion Assay
This protocol leverages a multi-color reporter to unequivocally distinguish true gene editing outcomes from background artifact, making it ideal for screening CRISPR efficacy [14] [32].
1. Principle: An eGFP-positive cell line is generated. A CRISPR-Cas9 ribonucleoprotein (RNP) complex is targeted to the eGFP locus. Repair via Non-Homologous End Joining (NHEJ) often disrupts the gene, leading to loss of fluorescence (knockout). Repair via Homology-Directed Repair (HDR) using a provided single-stranded oligodeoxynucleotide (ssODN) template introduces two specific point mutations, converting eGFP to BFP [32].
2. Materials:
- Cells: eGFP-positive HEK293T cells (generated via lentiviral transduction with pHAGE2-Ef1a-eGFP-IRES-PuroR) [32].
- Gene Editing Reagents:
- Equipment: Flow cytometer (e.g., BD FACS Canto II), electroporator or transfection reagent (e.g., ProDeliverIN CRISPR).
3. Procedure:
- Pre-production: Generate a stable, highly homogeneous eGFP-positive HEK293T cell line via lentiviral transduction and puromycin selection [32].
- Day 1: Seed the eGFP-positive cells.
- Day 2: Transfect or electroporate the cells with the pre-complexed Cas9 RNP and the HDR template ssODN.
- Days 5-7: Harvest cells and analyze fluorescence using a flow cytometer equipped with filters for GFP and BFP.
4. Analysis and Interpretation:
- The fluorescence profile will show four distinct populations, allowing for clear interpretation of editing outcomes [32]:
- GFP+ / BFP-: Non-edited cells.
- GFP- / BFP-: NHEJ-mediated knock-out.
- GFP- / BFP+: Successful HDR-mediated conversion.
- GFP+ / BFP+: Likely heterologous editing or artifact; exclude from analysis.
Diagram 2: CRISPR-Cas9 fluorescence conversion assay logic for unambiguous outcome tracking.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Addressing GFP Artifacts
| Reagent / Tool | Function / Application | Specifications / Examples |
|---|---|---|
| FPCountR | Converts plate reader RFU into absolute molecules of GFP per cell, enabling cross-lab comparisons [47]. | Open-access R package and wet lab protocol. Includes 'ECmax' assay for quantification without purification. |
| Fluorescence Conversion System | High-throughput readout for differentiating NHEJ vs. HDR CRISPR outcomes, bypassing promoterless GFP issues [32]. | Requires stable eGFP cell line, Cas9 RNP, and specific ssODN template to convert eGFP to BFP. |
| Marker-Free CRISPR Plasmids | Generates transgenic parasites/cells without integrated drug markers, reducing confounding promoter activity [48]. | Typically a two-plasmid system: one with Cas9/bsd, another with sgRNA/donor DNA and a positive-negative selectable marker (e.g., hdhfr-yfcu). |
| VISPR-online | Web-based tool for visualizing and exploring CRISPR screening data, aiding in QC and identifying potential artifacts [49]. | Supports outputs from MAGeCK, BAGEL, and JACKS. Allows interactive exploration of gene essentiality and gRNA read counts. |
The efficacy of CRISPR/Cas9 genome editing is fundamentally dependent on the careful design of the single-guide RNA (sgRNA). An optimal sgRNA ensures high on-target activity while minimizing off-target effects, which is crucial for generating reliable experimental results and for therapeutic safety. This application note provides a consolidated guide of modern tools, validation protocols, and strategies for enhancing sgRNA editing efficiency, with a specific focus on workflows that incorporate visual screening via GFP markers to streamline the isolation of successfully edited transformations.
Computational Tools for sgRNA Design and Off-Target Prediction
Selecting the right sgRNA begins with in silico analysis using specialized bioinformatics tools. These platforms assist researchers in choosing guides with high predicted on-target activity and low potential for off-target binding.
Table 1: Key Bioinformatics Tools for sgRNA Design and Analysis
| Tool Name | Primary Function | Key Features | Considerations |
|---|---|---|---|
| CRISPOR [50] | sgRNA design & off-target scoring | Robust design for several species, integrated off-target scoring, genomic locus visualization. | Versatile platform for comprehensive design. |
| CHOPCHOP [50] | sgRNA design & off-target scoring | Robust design for several species, integrated off-target scoring, genomic locus visualization. | Versatile platform for comprehensive design. |
| CCTop [13] | sgRNA design & off-target prediction | Used for guide design and to search for potential off-target sites. | Cited in optimization studies for human pluripotent stem cells. |
| CRISPRidentify [50] | CRISPR array identification | Employs machine learning to identify and distinguish genuine CRISPR arrays from false positives with high specificity. | Focuses on prokaryotic sequence analysis. |
| ICE (Inference of CRISPR Edits) [51] [52] | Analysis of editing efficiency | Free, fast analysis of Sanger sequencing data; provides editing efficiency and highlights off-target edits. | Used for post-experimental validation, not initial design. |
Beyond the initial design, it is critical to predict and minimize off-target activity. CRISPR off-target editing refers to non-specific activity of the Cas nuclease at sites other than the intended target, which can confound experimental results and pose significant safety risks in therapeutic applications [51]. Tools like CRISPOR and CHOPCHOP provide integrated off-target scoring, helping researchers select guides with low similarity to other genomic sites [50]. Strategies to minimize off-targets include choosing high-fidelity Cas variants, using chemically modified sgRNAs (e.g., with 2'-O-methyl analogs and 3' phosphorothioate bonds), and selecting guides with higher GC content, which stabilizes the DNA:RNA duplex [51].
Figure 1: A recommended workflow for the computational design and experimental selection of highly efficient sgRNAs.
Experimental Validation and Protocol for sgRNA Efficiency
In silico predictions require empirical validation. The following optimized protocol for human pluripotent stem cells (hPSCs), which can achieve indel efficiencies of 82-93% for single-gene knockouts, provides a robust framework for testing sgRNA activity [13].
Materials and Reagents
Table 2: Key Research Reagent Solutions for sgRNA Validation
| Reagent / Material | Function / Description | Example/Citation |
|---|---|---|
| iCas9 Cell Line | Doxycycline-inducible SpCas9-expressing cell line. Allows tunable nuclease expression. | hPSCs-iCas9 line [13] |
| Chemically Modified sgRNA (CSM-sgRNA) | Enhanced stability within cells via 2’-O-methyl-3'-thiophosphonoacetate modifications at both ends. | Synthesized by GenScript [13] |
| Validated Positive Control sgRNA | sgRNA with proven high editing efficiency to optimize workflow conditions. | Targets human genes like TRAC, RELA [52] |
| Nucleofection System | Method for efficient delivery of CRISPR components into cells. | 4D-Nucleofector (Lonza) using program CA137 [13] |
| Fluorescence Reporter (eGFP) | Transfection control to visually confirm delivery efficiency. | pKSE401G vector with 35S::sGFP [6] |
| ICE Analysis Tool | Software for analyzing Sanger sequencing data to determine editing efficiency. | Synthego's ICE (Inference of CRISPR Edits) [13] [51] |
Step-by-Step Protocol for Validating sgRNA Editing Efficiency
Cell Preparation and Transfection:
- Culture and passage your iCas9 cell line (e.g., H9 or H7 hPSCs) according to standard protocols [13].
- Induce Cas9 expression by adding doxycycline to the culture medium.
- Dissociate cells into a single-cell suspension and pellet them by centrifugation.
- For nucleofection, combine the cell pellet with a mixture containing your test sgRNA(s) and a positive control sgRNA (e.g., targeting a locus like ROSA26 for mouse cells or TRAC for human cells) [52]. A final amount of 5 µg of sgRNA for 8 × 10^5 cells is an effective ratio [13].
- Perform nucleofection using an optimized program (e.g., CA137 for hPSCs).
Include Critical Controls:
- Positive Editing Control: A validated sgRNA known to cut efficiently. This verifies your transfection and editing conditions are working [52].
- Negative Editing Controls: These establish a baseline for cellular phenotypes and confirm that observed effects are due to editing. Options include:
- Scramble sgRNA + Cas9: A non-targeting sgRNA.
- sgRNA Only: No Cas9 nuclease delivered.
- Cas9 Only: No guide RNA delivered [52].
- Transfection Control: Co-deliver a fluorescent reporter (e.g., GFP mRNA) to visually confirm and quantify delivery efficiency [52].
Post-Transfection Culture and Analysis:
- Allow transfected cells to recover and express the edited genome for 48-72 hours.
- Harvest cells and extract genomic DNA from the pooled population.
- Amplify the target genomic region by PCR and subject the product to Sanger sequencing.
- Analyze the sequencing chromatograms using the ICE tool or a similar algorithm (e.g., TIDE) to quantify the insertion/deletion (indel) efficiency [13].
Integrating Visual Markers for Efficient Isolation of Edited Clones
Visual markers dramatically streamline the identification and isolation of positive transformants and, crucially, the subsequent identification of transgene-free edited cells in later generations.
Application of GFP-Based Vectors
A modified CRISPR/Cas9 vector, pKSE401G, which incorporates a 35S::sGFP cassette, enables visual screening of primary transformants [6]. In practice, GFP-positive seeds or seedlings are easily identified under a fluorescence microscope, directly indicating the presence of the T-DNA (and thus the Cas9/sgRNA) [6]. This method has been successfully applied in Arabidopsis, B. napus, strawberry, and soybean, with mutation frequencies comparable to non-fluorescent vectors [6].
A Workflow for Isolating Transgene-Free Mutants Using Fluorescence
The following workflow, visualizable through fluorescence, allows for the efficient separation of edited from non-edited material without complex genotyping at every step.
Figure 2: A visual screening workflow for isolating transgene-free mutants in the T2 generation using a GFP marker.
- T1 Generation (Primary Transformants): After transformation with a visual marker vector (e.g., pKSE401G or ViMeRed vectors), identify and select primary transformants based on the presence of the fluorescent signal (e.g., GFP or DsRED2) [6] [53]. Confirm the presence of intended edits in these fluorescent-positive individuals via sequencing.
- T2 Generation (Isolating Transgene-Free): Self-pollinate the confirmed T1 plants and collect the seeds. In the T2 progeny, screen for individuals that lack the fluorescent signal. These GFP-negative plants have likely segregated away the T-DNA containing the Cas9/sgRNA and fluorescent marker genes, and are potential transgene-free mutants [6]. Genotype these candidates to confirm they retain the desired genetic edit.
This strategy has been proven effective, with one study reporting that 17.3% of T2 seedlings were GFP-negative (and thus Cas9-free) but still contained the desired mutation [6]. In maize, the ViMeBox toolbox uses seed-specific promoters (e.g., for aleurone or embryo) to drive DsRED2 expression, making Cas9-containing kernels visibly red and allowing for their easy separation from Cas9-free yellow kernels under natural light [53].
Optimizing sgRNA design is a multi-stage process that combines computational prediction with rigorous experimental validation. By leveraging modern bioinformatics tools, following optimized protocols for efficiency testing, and incorporating visual markers like GFP into the workflow, researchers can significantly enhance the efficiency and reliability of their CRISPR genome editing outcomes. This integrated approach not only accelerates the isolation of correctly edited clones but also facilitates the generation of transgene-free lines, which are crucial for both functional studies and agricultural applications.
In the field of genetic engineering, particularly for visual screening of CRISPR transformants with GFP markers, the efficiency with which genetic cargo is delivered into cells is a cornerstone of experimental success. Transfection efficiency directly impacts the robustness of data, the timeline of research, and the feasibility of advanced applications in drug development. The central choice researchers face is between viral and non-viral delivery methods, each with a distinct set of advantages, limitations, and optimal use cases. Viral vectors, such as lentiviruses and adeno-associated viruses (AAVs), are renowned for their high efficiency, especially in hard-to-transfect cells. In contrast, non-viral methods, including chemical reagents and electroporation, offer enhanced safety, simpler regulatory paths, and greater flexibility in cargo size. This application note provides a detailed, protocol-oriented comparison of these systems. It is structured within the context of CRISPR/GFP screening workflows, offering scientists a practical guide to selecting and optimizing the right delivery method for their specific research objectives, from basic science to therapeutic development.
Quantitative Comparison of Delivery Methods
Selecting the appropriate gene delivery method requires a clear understanding of key performance metrics. The following tables summarize the defining characteristics and efficiency data for common viral and non-viral vectors, providing a foundation for an informed choice.
Table 1: Key Characteristics of Viral and Non-Viral Delivery Methods [54] [55] [56]
| Feature | Viral Vectors (Lentivirus, AAV) | Non-Viral Methods (Lipids, Electroporation) |
|---|---|---|
| Typical Transfection Efficiency | High (can exceed 70-90% in permissive cells) [54] [57] | Variable; can be high in optimized systems (e.g., lipid-based) but often lower than viral in primary cells [58] |
| Cargo Capacity | Limited (~4.7 kb for AAV; ~8 kb for Lentivirus) [55] | Virtually unlimited [58] |
| Immunogenicity | Moderate to High [55] | Low [58] |
| Risk of Insertional Mutagenesis | Low with modern SIN designs, but still a consideration [54] | None [58] |
| Stability of Expression | Stable, long-term (integrating vectors) [54] | Transient (for most chemical methods) |
| Ease of Use & Production | Complex and time-consuming production process [59] | Simple, rapid protocol formulation [60] |
| Cost | High [58] | Relatively Low [58] |
| Best Suited For | Stable cell line generation, in vivo delivery, hard-to-transfect cells [54] [56] | Rapid knockout/knockin studies, delivery of large constructs, CRISPR RNP delivery [55] [61] |
Table 2: Impact of Transfection Parameters on Efficiency and Cell Health
This table outlines critical process parameters (CPPs) that require optimization to maximize efficiency while maintaining cell viability, a crucial balance in any transfection workflow. [54]
| Parameter | Impact on Transfection Efficiency | Impact on Cell Viability & Function | Optimization Consideration |
|---|---|---|---|
| Multiplicity of Infection (MOI) | Higher MOI generally increases transduction efficiency but can lead to saturation. [54] | High MOI can cause cytotoxicity and increase vector copy number (VCN), raising safety concerns. [54] | Titrate MOI to find the optimal balance for your cell type. Clinical programs often target VCN <5. [54] |
| Cell Health & Seeding Density | High viability and optimal density are prerequisites for high efficiency. Actively dividing cells are more susceptible. [54] | Poor starting viability and incorrect density lead to poor post-transfection recovery and function. [54] | Use cells in log-phase growth. Optimize seeding density for each cell type and vessel. |
| Enhancers (e.g., Polybrene, Peptides) | Can significantly boost viral transduction efficiency by promoting virus-cell attachment. [57] | Some enhancers (e.g., Polybrene) can be cytotoxic at high concentrations. [54] | Test different enhancers and concentrations. Transportan peptide shows efficacy with low cytotoxicity. [57] |
| Format of CRISPR Components (DNA, mRNA, RNP) | RNP format offers the fastest editing action and reduced off-target effects. [55] | DNA format leads to prolonged Cas9 expression, increasing off-target risk. RNP is rapidly degraded. [55] | RNP is preferred for precise editing. DNA is used for sustained selection pressure. |
The Scientist's Toolkit: Essential Reagents and Controls
A successful CRISPR transfection and screening experiment relies on more than just the delivery method. The following toolkit lists critical reagents, controls, and materials necessary for workflow optimization and validation.
Table 3: Research Reagent Solutions for CRISPR/GFP Workflows [62] [61] [52]
| Item | Function & Description | Example Use-Case |
|---|---|---|
| GFP-Expressing Viral Vectors | Used as a delivery optimization control. Fluorescence allows visual assessment of transduction efficiency and helps determine the optimal MOI. [62] | Co-transduce with your CRISPR-virus or use in a pilot experiment to image and quantify delivery success before your main experiment. |
| Validated Positive Control gRNA | A gRNA with known high editing efficiency against a standard gene (e.g., human TRAC, RELA). Verifies that transfection conditions are optimized for editing. [52] | Transfert alongside your experimental gRNA. High efficiency in the positive control confirms the workflow is functional. |
| Non-Targeting Negative Control gRNA | A gRNA with no known target in the genome. Establishes a baseline for cellular phenotype without CRISPR editing. [62] [52] | Crucial for distinguishing true editing-related phenotypes from non-specific effects of the transfection process itself. |
| Dual-Fluorescence Reporter Cell Line | A stable cell line (e.g., RFP-GFP) where successful CRISPR cutting repairs a broken GFP gene, turning cells GFP+. Enables quantification of functional CRISPR uptake. [61] | Use in a microplate reader assay to rapidly compare the functional efficiency of different transfection reagents or protocols. |
| Transfection Enhancers (e.g., Transportan) | Cell-penetrating peptides that, when co-administered, can enhance viral uptake via bystander macropinocytosis, boosting efficiency in hard-to-transfect cells. [57] | Simply mix Transportan peptide with your viral preparation (lentivirus or AAV) during incubation with cells to improve transduction. |
Experimental Protocols for Enhanced Efficiency
Protocol 1: Enhancing Viral Transduction with Transportan Peptide
This protocol describes a simple method to significantly improve lentiviral and AAV transduction efficiency, particularly in difficult-to-transfect cell lines and primary cells, using co-administration with Transportan (TP) peptide. [57]
Workflow Overview:
Materials:
- Transportan (TP) peptide (GWTLNSAGYLLGKINLKALAALAKKIL)
- GFP-expressing lentivirus or AAV
- Target cells (e.g., Raw264.7, primary macrophages, RPE cells)
- Complete cell culture medium
- Standard cell culture reagents and equipment
Step-by-Step Procedure:
- Cell Seeding: Plate the target cells at an appropriate density (e.g., 50-70% confluency) in a multi-well plate and allow them to adhere overnight.
- Mixture Preparation: Dilute the GFP-expressing viral vector in serum-free medium. To this dilution, add Transportan peptide to a final working concentration of 5-10 µM. Mix the solution gently by pipetting.
- Transduction: Remove the culture medium from the plated cells. Add the virus-TP mixture to the cells. Incubate the cells with this mixture for 48 hours at 37°C and 5% CO₂.
- Medium Replacement: After the incubation period, carefully remove the virus-TP-containing medium and replace it with fresh, complete culture medium.
- Analysis: Allow transgene expression to develop for an additional 24-48 hours. Analyze the transfection efficiency by quantifying the percentage of GFP-positive cells using flow cytometry or fluorescence microscopy.
Key Notes:
- This method is noted for its simplicity and low cytotoxicity. [57]
- The mechanism of enhancement is believed to involve TP-induced bystander uptake through macropinocytosis. [57]
Protocol 2: Optimizing Non-Viral Transfection of T Cells with PEI/DNA Nanoparticles
This protocol provides a tuned method for polyethylenimine (PEI)-mediated transfection of human T cells, a cell type critical for immunology and cell therapy research, which is notoriously difficult to transfect. [60]
Workflow Overview:
Materials:
- Linear or branched PEI transfection reagent
- Plasmid DNA (e.g., CRISPR-Cas9 plasmid with GFP marker)
- Human T cells (activated)
- RPMI 1640 medium
- Centrifuge tubes
Step-by-Step Procedure:
- Nanoparticle Formation: Prepare PEI/DNA nanoparticles at an optimal N/P ratio of 8 in a sterile tube. Vortex gently and incubate at room temperature for 15-20 minutes to allow for stable complex formation.
- Cell Preparation: Harvest activated T cells and seed them at a high density (e.g., 5-10 x 10^6 cells/mL) directly in centrifuge vials. This high local density dramatically improves transfection outcomes.
- "Reverse" Transfection: Add the pre-formed PEI/DNA nanoparticles directly to the cell pellet in the vial. Gently mix by tapping the tube. This "reverse" method (adding complexes to cells) was shown to increase uptake 20-fold compared to adding complexes to adhered cells in a plate. [60]
- Incubation and Feeding: Incubate the cell-nanoparticle mixture for a defined period (e.g., 4-6 hours). Then, carefully transfer the contents to a culture plate and add a sufficient volume of complete media to dilute the complexes and support cell growth.
- Analysis: After 48-72 hours, assess transfection efficiency by measuring GFP expression via flow cytometry. For CRISPR experiments, genomic DNA should be harvested and analyzed by T7E1 assay or next-generation sequencing to determine editing efficiency.
Key Notes:
- Further modulation of cellular physiology using hypotonic extracellular media at pH 9.0 can dramatically enhance transfection rates while maintaining minimal cytotoxicity. [60]
- Always include the positive and negative controls listed in Table 3 to validate the experiment.
Visual Screening and Validation of CRISPR Transformants
The ultimate validation of a successful transfection is the intended genetic modification. Using GFP markers and fluorescent reporters enables rapid and visual screening of CRISPR transformants.
Application of the Dual-Fluorescence Reporter System: A stably integrated RFP-GFP reporter system is a powerful tool for quantifying the delivery of functional CRISPR-Cas9 components. [61] In this system, cells constitutively express mRFP, which serves as a transfection control and a marker for selecting reporter-positive cells. The GFP gene is out-of-frame and only becomes functional upon CRISPR-Cas9-induced double-strand break and error-prone non-homologous end joining (NHEJ) repair that fixes the reading frame.
Screening Protocol:
- Transfection: Transfect the stable reporter cell line with your CRISPR-Cas9 components (e.g., as RNP complex).
- Incubation: Allow 48-72 hours for expression and editing to occur.
- Analysis: Use flow cytometry or a microplate reader to detect and quantify the double-positive (RFP+GFP+) cell population. The percentage of double-positive cells directly represents the percentage of cells with successful, functional CRISPR editing. [61]
- Enrichment: The double-positive cell population can be isolated using fluorescence-activated cell sorting (FACS) to generate a highly enriched pool of knock-out cells for downstream functional assays.
The choice between viral and non-viral delivery methods is not a matter of declaring one superior to the other, but rather of matching the method's strengths to the experiment's requirements. Viral methods offer high efficiency and stability for long-term studies and challenging cell types, while non-viral methods provide a safer, more flexible, and rapid solution for many CRISPR applications, particularly with the advent of RNP delivery. As demonstrated in the protocols, efficiency for both systems can be significantly enhanced through strategic optimization, such as the use of peptide enhancers like Transportan for viral vectors or refined nanoparticle formulation and "reverse" transfection for non-viral methods. By leveraging the quantitative comparisons, essential toolkits, and detailed protocols outlined in this application note, researchers can systematically improve their transfection workflows. This will lead to more reliable and efficient visual screening of CRISPR transformants, accelerating the pace of discovery and therapeutic development.
In the field of visual screening of CRISPR transformants with GFP markers, understanding the inherent DNA repair capacities of different cell lines is paramount for experimental design and data interpretation. CRISPR/Cas9-mediated genome editing relies entirely on the cellular machinery that repairs DNA double-strand breaks (DSBs), primarily through non-homologous end joining (NHEJ) or homology-directed repair (HDR) pathways [32] [63]. The efficiency and fidelity of CRISPR editing outcomes are profoundly influenced by which of these pathways a cell utilizes, and this preference varies significantly across cell types due to differences in expression of repair proteins, cell cycle distribution, and metabolic states [63]. The use of GFP-based reporters provides a powerful visual tool to quantify these repair outcomes, but researchers must account for cell line-specific repair characteristics to draw valid conclusions.
The fundamental principle behind GFP-based repair reporting systems involves engineering a detectable fluorescent signal change upon successful DNA repair. A common approach involves mutating specific amino acids in the eGFP sequence through single nucleotide polymorphisms to shift fluorescence from green to blue, while insertions or deletions at this locus lead to complete loss of fluorescence [32]. This system enables simultaneous differentiation between HDR and NHEJ activities, providing a quantitative measure of how different cell lines handle the same DNA lesion. However, the baseline drift in DNA repair capabilities across cell lines can significantly impact the results, necessitating careful experimental design and interpretation [63].
DNA Repair Pathways: Mechanisms and Clinical Relevance
Historical Context and Molecular Mechanisms
The DNA damage response (DDR) encompasses a complex network of pathways that preserve genome integrity, with failures in these systems leading to human diseases including cancer and premature aging [63] [64]. The discovery of DNA repair mechanisms dates back to the 1920s with Hermann Muller's demonstration that X-rays induce genetic mutations, followed by the formal introduction of "DNA repair" terminology in 1964 with the discovery of "Dark Repair" and photo reactivating "repair-replication" mechanisms [63]. In 2015, the Nobel Prize in Chemistry recognized Tomas Lindahl, Paul Modrich, and Aziz Sancar for their pioneering work in mapping DNA repair processes at the molecular level.
Mammalian cells have evolved multiple sophisticated pathways to repair different types of DNA damage. The two primary pathways relevant to CRISPR/Cas9 editing are:
Non-Homologous End Joining (NHEJ): The dominant pathway in most mammalian cells, NHEJ directly ligates broken DNA ends with minimal regard for sequence conservation, often resulting in small insertions or deletions (indels) [32]. This pathway operates throughout the cell cycle and is error-prone, making it ideal for gene knockout strategies.
Homology-Directed Repair (HDR): A more precise mechanism that requires a DNA template for repair, HDR is restricted primarily to the S and G2 phases of the cell cycle when sister chromatids are available [32] [65]. HDR occurs less frequently than NHEJ but enables precise genetic modifications, including specific base changes or insertions.
The balance between these pathways varies across cell types due to differential expression of key repair proteins, cell cycle distribution, and metabolic states, creating a "DNA damage baseline drift" that significantly impacts CRISPR editing outcomes [63].
DNA Repair Pathway Diagram
Diagram Title: DNA Double-Strand Break Repair Pathways
This diagram illustrates the two main DNA repair pathways relevant to CRISPR/Cas9 editing: Non-Homologous End Joining (NHEJ) and Homology-Directed Repair (HDR). The cell cycle phase significantly influences which pathway is utilized, with NHEJ dominating in G0/G1 phases and HDR being restricted primarily to S/G2 phases. Key proteins involved in each step are indicated, highlighting potential bottlenecks where cell line variations can impact editing outcomes.
Cell Line-Specific DNA Repair Characteristics
Quantitative Comparison of Repair Efficiencies
Different cell lines exhibit substantial variation in their DNA repair capacities due to differences in gene expression profiles, genetic backgrounds, and physiological states. The table below summarizes documented repair characteristics for commonly used cell lines in CRISPR screening:
Table 1: DNA Repair Characteristics of Common Cell Lines in CRISPR Screening
| Cell Line | Origin | NHEJ Efficiency | HDR Efficiency | Reporter Validation | Key Considerations |
|---|---|---|---|---|---|
| HEK293T | Human Embryonic Kidney | High [32] | Moderate [32] | eGFP-BFP Conversion [32] | Robust growth, high transfection efficiency, suitable for initial optimization |
| HepG2 | Human Hepatocellular Carcinoma | Moderate [32] | Low [65] | Promoterless EGFP [65] | Metabolic competence, relevant for toxicology studies |
| RPE-1 | Human Retinal Pigment Epithelium | Well-characterized [64] | Well-characterized [64] | SPIDR Library Screening [64] | Karyotypically normal, TP53 wildtype (unless modified) |
| Porcine Fetal Fibroblasts (PFF) | Porcine Fetal Tissue | Variable [65] | Variable [65] | Promoterless EGFP [65] | Species-specific considerations, agricultural research |
| HeLa | Human Cervical Cancer | High [66] | Low [66] | Promoterless GFP [66] | HPV-transformed, defective p53 pathway |
| IMR90 | Human Fetal Lung Fibroblasts | Documented [32] | Documented [32] | eGFP-BFP Conversion [32] | Finite lifespan, primary cell characteristics |
Experimental Evidence of Cell Line Variability
Substantial evidence demonstrates the impact of cell line selection on editing outcomes. A comprehensive study interrogating synthetic lethality in the DNA damage response revealed that genetic interactions between DDR genes can vary significantly across cell lines [64]. When the same dual sgRNA screening was performed in RPE-1, HeLa S3, and K562 cells, researchers found both shared and cell-type-specific synthetic lethal interactions, highlighting the context-dependent nature of DNA repair pathway utilization [64].
The accuracy of reporter systems themselves can also vary by cell type. A validation study examining promoterless EGFP reporters found unexpectedly high background EGFP expression when the same porcine GAPDH-targeting construct was transfected into various cell lines, with significantly different expression levels observed in HepG2, HepaRG, 293T, and CHO-K1 cells [65]. This cell-type-dependent background noise can compromise the accuracy of HDR efficiency measurements if not properly characterized for each cell model.
Furthermore, the same CRISPR-mediated DSBs can trigger different downstream consequences across cell types. In HeLa and RKO cells engineered with a promoterless GFP construct, Cas9-induced DSBs activated DNA damage response, reduced cell viability, and increased mortality, but the magnitude of these effects differed between lines [66]. These differences extended to morphological changes, with variations in cell size, multinucleation, and cGAS-positive micronuclei accumulation observed between cell types following equivalent DNA damage [66].
Experimental Workflow for Cell Line Evaluation
Comprehensive Evaluation Protocol
Diagram Title: Cell Line Repair Capacity Evaluation Workflow
This workflow outlines a systematic approach for characterizing the DNA repair capacities of different cell lines using GFP-based reporter systems. The process begins with establishing stable reporter lines, followed by CRISPR-mediated targeting of the fluorescent protein locus, quantitative analysis of repair outcomes, and validation using orthogonal methods. Optimization cycles may be necessary for cell lines with initially low editing efficiencies.
Detailed Methodology for Repair Capacity Assessment
Protocol: Evaluation of Cell Line-Specific DNA Repair Capacities Using eGFP-BFP Conversion System
Materials and Equipment:
- eGFP-positive cell lines (generated via lentiviral transduction) [32]
- SpCas9-NLS protein [32]
- sgRNA targeting eGFP locus: GCUGAAGCACUGCACGCCGU [32]
- HDR template containing BFP mutations [32]
- Transfection reagent (e.g., Polyethylenimine or ProDeliverIN CRISPR) [32]
- Complete cell culture medium (DMEM + 10% FBS) [32]
- Flow cytometer (e.g., BD FACS Canto II) [32]
- Software for data analysis (FlowLogic, GraphPad Prism) [32]
Step-by-Step Procedure:
Cell Line Preparation and Culture
- Maintain all cell lines in complete culture medium at 37°C with 5% CO₂ [32].
- For each cell line, establish eGFP-positive cells via lentiviral transduction using pHAGE2-Ef1a-eGFP-IRES-PuroR plasmid and select with puromycin (2 μg/mL) [32].
- Culture cells for at least one week before experimentation to ensure robust growth and stable eGFP expression.
CRISPR/Cas9 Transfection
- Seed eGFP-positive cells at appropriate density (e.g., 1×10⁵ cells/well in 12-well plates) 24 hours before transfection [32].
- Prepare ribonucleoprotein (RNP) complexes by incubating 2 μg SpCas9-NLS with 1 μg sgRNA for 10 minutes at room temperature [32].
- For HDR assessment, include 1 μg of single-stranded oligo deoxynucleotide (ssODN) HDR template containing BFP-converting mutations: caagctgcccgtgccctggcccaccctcgtgaccaccctgAGCCACggcgtgcagtgcttcagccgctaccccgaccacatgaagc [32].
- Transfert using appropriate reagent following manufacturer's protocols (e.g., 4 μL ProDeliverIN CRISPR per μg DNA) [32].
Post-transfection Handling and Analysis
- Incubate cells for 48-72 hours to allow for repair and expression of editing outcomes [32].
- Harvest cells using trypsin-EDTA and resuspend in PBS containing 2% FBS [32].
- Analyze fluorescence using flow cytometry with appropriate filters for eGFP (excitation: 488 nm, emission: 507 nm) and BFP (excitation: 402 nm, emission: 457 nm) [32].
- Include untransfected eGFP-positive cells as negative control and single-color compensation controls.
Data Processing and Interpretation
- Gate on viable cells using forward and side scatter profiles [32].
- Quantify the percentage of BFP-positive cells (successful HDR) and non-fluorescent cells (NHEJ-induced knockout) [32].
- Calculate HDR efficiency as: (Number of BFP-positive cells / Total viable cells) × 100 [32].
- Calculate NHEJ efficiency as: (Number of non-fluorescent cells / Total viable cells) × 100 [32].
- Account for background fluorescence and autofluorescence using untransfected controls.
Research Reagent Solutions for DNA Repair Studies
Table 2: Essential Research Reagents for DNA Repair Capacity Evaluation
| Reagent Category | Specific Examples | Function in Assay | Application Notes |
|---|---|---|---|
| Fluorescent Reporters | pHAGE2-Ef1a-eGFP-IRES-PuroR [32] | Establishes baseline fluorescence for repair tracking | Enables visual tracking of repair outcomes; puromycin resistance allows selection of stable lines |
| Editing Enzymes | SpCas9-NLS [32] | Induces targeted double-strand breaks | Nuclear localization signal (NLS) ensures proper cellular localization |
| Repair Templates | ssODN with BFP-converting mutations [32] | Provides template for homology-directed repair | Contains specific nucleotide changes to convert eGFP to BFP; often includes PAM site mutation to prevent re-cutting |
| Delivery Vehicles | ProDeliverIN CRISPR [32], Polyethylenimine (PEI) [32] | Facilitates intracellular delivery of editing components | Chemical transfection reagents suitable for difficult-to-transfect cell lines |
| Selection Agents | Puromycin [32] | Selects for successfully transduced cells | Concentration must be optimized for each cell line (typically 1-5 μg/mL) |
| Analysis Tools | Flow cytometer with 402nm and 488nm lasers [32] | Quantifies fluorescence changes | Essential for accurate quantification of eGFP to BFP conversion and knockout efficiency |
Technical Considerations and Optimization Strategies
Addressing Cell Line-Specific Challenges
When working with diverse cell lines, several technical challenges may arise that require specific optimization strategies:
Low Transfection Efficiency: For cell lines with inherently low transfection efficiency (e.g., primary cells, some stem cells), consider alternative delivery methods such as electroporation or viral delivery. The choice of transfection reagent significantly impacts efficiency and should be optimized for each cell type [32].
Variable HDR Efficiency: Cell lines with inherently low HDR efficiency may benefit from synchronization in S/G2 phase or the use of small molecule enhancers such as RS-1, L755507, or SCR7 [65]. Additionally, optimizing the design and length of homology arms in repair templates can improve HDR rates in recalcitrant cell lines.
Background Fluorescence Issues: As observed in promoterless EGFP systems, some cell lines exhibit unexpected background expression that can confound results [65]. Conduct thorough validation with transfection controls (donor template alone without Cas9/sgRNA) to establish baseline signals for each cell line.
Cell Line-Specific Toxicity: Certain cell lines may exhibit heightened sensitivity to DNA damage, leading to substantial cell death following CRISPR editing [66]. Titrating Cas9 concentration and optimizing delivery methods can mitigate this toxicity while maintaining efficient editing.
Validation and Quality Control
Robust validation of DNA repair capacity measurements requires multiple orthogonal approaches:
Sequencing Verification: Confirm editing outcomes by Sanger sequencing or next-generation sequencing of target loci across cell lines to validate that fluorescence patterns accurately represent intended genetic modifications [32].
Functional Confirmation: Implement functional assays specific to the DNA repair pathways being studied, such as immunofluorescence for repair protein recruitment or Western blotting for phosphorylation of DNA damage sensors (e.g., γH2AX, p53) [66].
Multiple Reporter Systems: Where possible, employ complementary reporter systems to cross-validate results, such as using both eGFP-BFP conversion and promoterless EGFP knock-in approaches in parallel [32] [65].
Proper characterization of cell line-specific DNA repair capacities enables more accurate experimental design, appropriate model selection for specific research questions, and more reliable interpretation of CRISPR screening results in the context of visual GFP marker systems.
In the context of visual screening for CRISPR transformants using GFP markers, achieving a high signal-to-noise ratio (SNR) is paramount for accurately identifying successfully edited events. Background fluorescence can obscure true positive signals, leading to both false positives and false negatives, thereby compromising screening efficiency and data reliability. This application note details proven protocols and strategies for optimizing SNR by systematically reducing background fluorescence, enabling researchers to obtain cleaner and more interpretable results in CRISPR-Cas9 experiments involving fluorescent reporters.
The following table summarizes key metrics and strategies for optimizing signal-to-noise ratios in fluorescent-based screening, as identified from current research.
Table 1: Quantitative Data and Strategies for Fluorescence Signal-to-Noise Optimization
| Factor | Reported Metric/Strategy | Impact on Signal-to-Noise | Source/Context |
|---|---|---|---|
| Reporter Type | Endogenous NRX-1::Skylan-S vs. overexpressed mig-13p::CLA-1::GFP | SNR of 4.09 ± 0.23 vs. 18.5 ± 1.9 [67] | Highlights challenge of dimmer signals from endogenous tagging. [67] |
| Visual Marker | Use of dsRED2/tdTomato | Enables naked-eye screening without instruments; tdTomato produces pink tissue under white light. [68] [69] | Reduces background from instrument autofluorescence; simplifies and accelerates screening. [68] [69] |
| Thresholding Algorithm | Bradley's local means method | More robust against varying background and low SNR compared to global thresholding. [67] | Improves accuracy of puncta detection and quantification in image analysis. [67] |
| Cell Coverage | High power maintained even at low cell coverage when using Bayesian analysis (Waterbear) [70] | Allows reduction in cell numbers while maintaining statistical power in FACS screens. [70] | |
| Promoter for Cas9 | WUS promoter vs. EC1.2 promoter | Editing efficiency increased from ~38.5% to ~66.7% in T1 generation [69] | Higher efficiency reduces screening burden and background from unsuccessful editing. [69] |
Experimental Protocols
Protocol 1: Two-Step CRISPR-Cas9 for Precise TE Deletion with Fluorescent Marker
This protocol is designed to replace a transposable element (TE) with a fluorescent marker (DsRed) via homology-directed repair (HDR) in Drosophila melanogaster, allowing for visual screening of precise edits while preserving the genetic background [71].
Before You Begin:
- Expand the Drosophila natural population for 2–4 weeks to have enough flies for microinjection [71].
- Prepare microinjection setup or contact a commercial injection service [71].
Steps:
- sgRNA and Repair Plasmid Construction:
- Design two sgRNAs (sgRNA1, sgRNA2) flanking the target TE using a tool like Target Finder [71].
- Clone sgRNAs into the pCFD5 plasmid [71].
- Synthesize homology arms (~500bp) from the genomic regions upstream and downstream of the TE. Introduce silent mutations in the PAM sequences to prevent re-cleavage [71].
- Assemble a repair plasmid (e.g., using pHD-ScarlessDsRed) containing the homology arms and the DsRed fluorescent marker cassette via HiFi DNA assembly [71].
Microinjection and Screening (Step 1 - TE Replacement):
- Co-inject the pCFD5-sgRNA plasmid and the repair plasmid into embryos of the target Drosophila strain [71].
- Cross the injected individuals (G0) and screen their progeny (G1) under a fluorescence microscope for individuals expressing DsRed, which indicate successful HDR [71].
- Establish stable mutant lines from DsRed-positive flies [71].
Marker Excision (Step 2 - Creating a Scarless Deletion):
- Design two new sgRNAs (sgRNA3, sgRNA4) targeting sequences immediately flanking the inserted DsRed cassette [71].
- Clone these into a new pCFD5 plasmid and co-inject with a repair plasmid containing only the original homology arms (lacking the TE and DsRed) into embryos from the DsRed-positive line [71].
- Screen the progeny for the loss of DsRed fluorescence, indicating precise excision of the marker and the TE, leaving a clean deletion [71].
- Confirm the final genotype by PCR and sequencing [71].
Protocol 2: Rapid Screening of CRISPR Editing Outcomes via eGFP to BFP Conversion
This cell-based protocol uses a fluorescent protein conversion assay to simultaneously quantify two major DNA repair outcomes: Non-Homologous End Joining (NHEJ) and Homology-Directed Repair (HDR) [32].
Before You Begin:
- Generate eGFP-positive HEK293T cells via lentiviral transduction using the pHAGE2-Ef1a-eGFP-IRES-PuroR plasmid and select with puromycin [32].
- Culture cells in complete DMEM medium with 10% FBS [32].
Steps:
- Transfection with CRISPR Components:
- Design a sgRNA to target the eGFP sequence. The repair template is an ssODN that introduces two specific point mutations to convert eGFP into BFP [32].
- One day before transfection, seed eGFP-positive HEK293T cells in a multi-well plate.
- Transfect the cells with a complex of SpCas9 protein, the anti-eGFP sgRNA, and the HDR template ssODN using a transfection reagent like Polyethylenimine (PEI) or ProDeliverIN CRISPR [32].
Post-Transfection Cell Handling and Analysis:
- Incubate transfected cells for 48-72 hours to allow for expression of editing outcomes [32].
- Harvest cells, wash with PBS, and resuspend in a FACS-compatible buffer, optionally fixing with paraformaldehyde for preservation [32].
- Analyze cell fluorescence using a flow cytometer (e.g., BD FACS Canto II) with appropriate filter sets for eGFP (FITC channel) and BFP (DAPI or Pacific Blue channel) [32].
Data Interpretation:
Essential Research Reagent Solutions
The following table catalogs key reagents and their functions for implementing the described fluorescence-based CRISPR screening protocols.
Table 2: Key Research Reagents for Fluorescence-Based CRISPR Screening
| Reagent/Tool | Function/Application | Protocol/Context |
|---|---|---|
| pCFD5 Plasmid | Cloning vector for sgRNA expression in Drosophila [71]. | Two-step TE Deletion [71] |
| pHD-ScarlessDsRed | Repair template plasmid for HDR, contains DsRed marker and homology arms [71]. | Two-step TE Deletion [71] |
| pnos-Cas9-nos | Cas9 expression vector for Drosophila [71]. | Two-step TE Deletion [71] |
| SpCas9-NLS | Purified Cas9 protein for formation of Ribonucleoprotein (RNP) complexes for direct delivery [32]. | eGFP to BFP Conversion [32] |
| HDR Template ssODN | Single-stranded DNA oligonucleotide encoding desired mutations (e.g., eGFP>BFP) and a mutated PAM to prevent re-cleavage [32]. | eGFP to BFP Conversion [32] |
| Polyethylenimine (PEI) | A transfection reagent for delivering CRISPR components into cells [32]. | eGFP to BFP Conversion [32] |
| dsRED2 | A red fluorescent protein visible to the naked eye in certain tissues, used as a visual screening marker without specialized equipment [69]. | Visual Screening [69] |
| WUS promoter | A promoter that drives Cas9 expression in meristematic cells, shown to increase editing efficiency in plants [69]. | Visual Screening [69] |
| Waterbear Software | A Bayesian computational tool for robust analysis of FACS-based CRISPR screen data, especially with limited replicates or cells [70]. | Data Analysis [70] |
| WormSNAP Software | A no-code, stand-alone tool for unbiased detection and characterization of fluorescent puncta in 2D images, using adaptive thresholding [67]. | Image Analysis [67] |
Workflow and Pathway Visualizations
The following diagrams outline the core experimental and analytical processes for optimizing signal-to-noise in fluorescence-based CRISPR screening.
Visual Screening and Analysis Workflow
Signal-to-Noise Optimization Strategies
Low knockout efficiency remains a significant bottleneck in CRISPR-Cas9 experiments, often leading to variable results and failed experiments. Within the broader context of visual screening for CRISPR transformants using GFP and other fluorescent markers, systematic troubleshooting becomes paramount for research reliability. This protocol provides a comprehensive, step-by-step framework for diagnosing and resolving the most common causes of low knockout efficiency, integrating both established and emerging visual screening methodologies to accelerate successful genome editing.
The persistent challenge of inefficient gene editing affects functional genomics studies and therapeutic applications alike. By implementing the systematic approach outlined below, researchers can significantly improve their editing outcomes, leveraging visual reporters not only for screening but also for rapid efficiency assessment.
Understanding and Measuring Knockout Efficiency
Knockout efficiency refers to the percentage of cells in a population that contain successful disruption of the target gene, typically through frameshift mutations or deletions introduced by non-homologous end joining (NHEJ) [72]. Accurate measurement of this efficiency is fundamental to troubleshooting.
Key Measurement Methods:
- Next-Generation Sequencing (NGS): Considered the gold standard, NGS provides comprehensive data on editing spectra and frequency with high sensitivity, though it requires significant resources and bioinformatics support [73].
- Inference of CRISPR Edits (ICE): This accessible tool analyzes Sanger sequencing data to determine indel frequencies and editing patterns, demonstrating strong correlation (R² = 0.96) with NGS results [73].
- Tracking of Indels by Decomposition (TIDE): Another Sanger sequencing analysis method that quantifies editing efficiency but has limitations in detecting complex indel patterns [73].
- T7 Endonuclease I (T7E1) Assay: A rapid, cost-effective method that detects mismatches in heteroduplex DNA but provides limited quantitative data and no sequence-level information [73].
Table 1: Comparison of CRISPR Analysis Methods
| Method | Sensitivity | Information Depth | Cost | Throughput | Best Use Cases |
|---|---|---|---|---|---|
| NGS | High | Complete sequence data | High | High | Publication-quality data, novel editing characterization |
| ICE | Medium-High | Indel types and frequencies | Low-Medium | Medium | Routine validation, optimization experiments |
| TIDE | Medium | Simplified indel profiles | Low-Medium | Medium | Initial efficiency assessment |
| T7E1 Assay | Low | Presence/absence of editing | Low | High | Quick confirmation during optimization |
Common Causes and Solutions for Low Knockout Efficiency
Suboptimal sgRNA Design
sgRNA design fundamentally influences cleavage efficiency and specificity. Poorly designed guides result in inadequate target binding and reduced knockout rates [72].
Solutions:
- Utilize bioinformatics tools like CRISPR Design Tool or Benchling to predict optimal sgRNAs with high on-target activity and minimal off-target potential [72].
- Test 3-5 different sgRNAs per gene target to identify the most effective sequence [72].
- Consider GC content, which significantly impacts efficiency. Research in grape systems demonstrated that sgRNAs with 65% GC content yielded highest editing efficiency compared to lower GC alternatives [74].
- Avoid secondary structures that may interfere with sgRNA-Cas9 binding.
Inefficient Delivery and Expression
Successful delivery of CRISPR components remains a critical hurdle, particularly in difficult-to-transfect cell types.
Solutions:
- Optimize Transfection Method: Lipid-based transfection reagents (e.g., DharmaFECT, Lipofectamine 3000) work well for many mammalian cells, while electroporation may be superior for challenging cell types [72].
- Utilize Stable Cas9 Cell Lines: Stably expressing Cas9 cell lines eliminate transfection variability and provide consistent nuclease expression, significantly enhancing reproducibility [72].
- Implement Inducible Systems: Doxycycline-inducible Cas9 systems (iCas9) in human pluripotent stem cells have achieved INDEL efficiencies of 82-93% for single-gene knockouts after systematic optimization [13].
- Employ Modified sgRNA: Chemically synthesized and modified (CSM) sgRNAs with 2'-O-methyl-3'-thiophosphonoacetate modifications at both ends demonstrate enhanced stability within cells [13].
Cell Line-Specific Variations
Different cell lines exhibit variable responses to CRISPR editing due to inherent biological differences.
Solutions:
- Account for DNA Repair Variations: Certain cell lines (e.g., HeLa) possess highly efficient DNA repair mechanisms that can counteract Cas9-induced breaks [72].
- Optimize Cell-Specific Parameters: Research demonstrates significant efficiency differences between varieties, with '41B' grape cells showing higher editing efficiency than 'Chardonnay' under identical conditions [74].
- Adjust Cell Culture Conditions: Ensure optimal cell health and proliferation status at transfection, as this significantly impacts editing outcomes.
Off-Target Effects
Unexpected cleavage at off-target sites can divert editing resources from the intended target and complicate results interpretation [75].
Solutions:
- Utilize Specificity-Enhanced Cas9 Variants: Engineered Cas9 versions with improved fidelity reduce off-target cleavage while maintaining on-target activity.
- Employ Computational Prediction: Tools that identify potential off-target sites help in guide selection and post-editing analysis.
- Implement Unbiased Detection Methods: Techniques like GUIDE-seq, BLESS, and Digenome-seq provide genome-wide off-target profiling without predicated sites [75].
Visual Screening Systems for Rapid Efficiency Assessment
The integration of visual markers provides powerful tools for rapid identification of successfully edited cells, significantly streamlining the screening process.
Fluorescent Protein Reporters
Traditional fluorescent proteins remain invaluable for visual screening:
- Diverse Color Palette: Eleven fluorescent proteins spanning red, green, yellow, and cyan spectra have been successfully expressed in cotton, with tdTomato-transgenic tissue appearing pink under white light [68].
- Stable Expression: Nine tested FPs (mCherry, tdTomato, sfGFP, Clover, EYFP, YPet, mVenus, mCerulean, and ECFP) showed stable, intense expression in transgenic callus and embryo tissues, with inheritance in mature organs [68].
- Subcellular Localization Markers: FP-labeled markers enable precise localization to seven subcellular compartments: plasma membrane, endoplasmic reticulum, tonoplast, mitochondrion, plastid, Golgi apparatus, and peroxisome [68].
Native Visual Screening Reporters (NVSR)
Endogenous visual markers overcome limitations associated with traditional fluorescent reporters:
- Plant-Based Systems: In strawberry, FveMYB10 overexpression under tissue-specific promoters induced visible red pigmentation in transgenic calli, with 97% of red calli testing positive for Cas9 [8].
- Temporal Regulation: Promoters driving NVSR can be temporally regulated, allowing initial visual identification followed by pigment disappearance at later developmental stages, minimizing interference with plant development [8].
- Cost-Effectiveness: NVSR systems enable naked-eye screening without specialized equipment or costly substrates, significantly reducing screening costs and effort [8].
Experimental Protocols for Efficiency Optimization
Protocol: High-Efficiency Knockout in Human Pluripotent Stem Cells
This optimized protocol achieves 82-93% INDEL efficiency in hPSCs [13]:
Materials:
- Doxycycline-inducible Cas9 hPSC line (hPSCs-iCas9)
- Chemically synthesized and modified sgRNA (CSM-sgRNA)
- Nucleofection system (Lonza 4D-Nucleofector)
- P3 Primary Cell Nucleofection Kit
- Doxycycline
Procedure:
- Culture hPSCs-iCas9 in Pluripotency Growth Master 1 Medium on Matrigel-coated plates.
- Induce Cas9 expression with 1-2 μg/mL doxycycline 24 hours before nucleofection.
- Dissociate cells with 0.5 mM EDTA and pellet by centrifugation at 250g for 5 minutes.
- Resuspend 8 × 10^5 cells in nucleofection buffer with 5μg CSM-sgRNA.
- Electroporate using CA137 program on Lonza Nucleofector.
- Repeat nucleofection after 3 days using identical parameters.
- Analyze editing efficiency 72 hours post-second nucleofection using ICE analysis of Sanger sequencing data.
Troubleshooting Notes:
- Cell density and viability at nucleofection critically impact efficiency.
- CSM-sgRNA outperforms in vitro transcribed sgRNA due to enhanced stability.
- Multiple nucleofections significantly increase editing rates in pooled cells.
Protocol: Visual Screening-Based Selection in Plants
This protocol leverages visual markers for efficient identification of CRISPR-edited plant materials [8] [68]:
Materials:
- Agrobacterium strain harboring CRISPR/NVSR construct
- Plant explants for transformation
- Tissue-specific promoter-driven FveMYB10 or FP construct
- Selective antibiotics appropriate for the transformation system
Procedure:
- Transform plant explants via Agrobacterium-mediated transformation.
- Culture transformed tissues under appropriate selection pressure.
- Screen for visual marker expression (red pigmentation for NVSR, fluorescence for FPs).
- Isolate positive transformants based on visual signal.
- Confirm editing via molecular analysis (PCR, sequencing).
- Regenerate whole plants from confirmed edited tissues.
Validation Data:
- In strawberry, 97% (30/31) of red pigmented calli were Cas9-positive, versus only 36% (10/28) of non-pigmented calli [8].
- Mutation rates of 100% for sgRNA1 and 73.3% for sgRNA2 were achieved in red calli lines [8].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Optimizing CRISPR Knockout Efficiency
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| sgRNA Design Tools | Benchling, CRISPR Design Tool, CCTop | Predict optimal sgRNAs with high on-target activity and minimal off-target effects; Benchling shows superior accuracy in experimental validation [72] [13]. |
| Analysis Software | ICE (Inference of CRISPR Edits), TIDE, NGS pipelines | Quantify editing efficiency from sequencing data; ICE provides NGS-comparable accuracy from Sanger data at lower cost [73] [13]. |
| Delivery Reagents | Lipid-based transfection reagents (DharmaFECT, Lipofectamine), Electroporation systems | Deliver CRISPR components into cells; choice depends on cell type with electroporation superior for challenging cells [72]. |
| Visual Reporters | FPs (tdTomato, mCherry, sfGFP), Native reporters (FveMYB10) | Enable rapid visual screening of transformants; NVSR systems allow naked-eye identification without specialized equipment [8] [68]. |
| Specialized Cell Lines | Stably expressing Cas9 lines, Inducible Cas9 systems (iCas9) | Provide consistent Cas9 expression, eliminating transfection variability; iCas9 systems enable temporal control [72] [13]. |
| Modified sgRNA | CSM-sgRNA (2'-O-methyl-3'-thiophosphonoacetate modified) | Enhance sgRNA stability and editing efficiency through chemical modifications that reduce degradation [13]. |
Workflow Diagram for Systematic Troubleshooting
The following diagram outlines a systematic approach to diagnosing and resolving low knockout efficiency issues:
Systematic Troubleshooting Workflow for CRISPR Knockout Efficiency
Successful CRISPR knockout experiments require integrated optimization across multiple parameters, from sgRNA design to delivery methods and appropriate screening approaches. By implementing the systematic troubleshooting framework outlined here and leveraging visual screening technologies, researchers can significantly improve their editing efficiencies. The consistent application of validated measurement tools like ICE analysis, combined with the rapid screening capabilities of visual reporter systems, provides a robust pathway to reliable genome editing outcomes. As CRISPR technology continues to evolve, these foundational principles will remain essential for maximizing experimental success across diverse biological systems and applications.
Beyond GFP: Validating CRISPR Edits and Comparing Methodologies
The application of CRISPR/Cas9 technology for genome editing has revolutionized functional genomics, enabling precise modifications in cellular DNA. A typical CRISPR experiment involves designing guide RNAs (gRNAs), selecting the appropriate Cas nuclease, introducing these components into cells, and ultimately validating the resulting genetic edits [76]. Within the specific context of research focused on visual screening of CRISPR transformants with GFP markers, validation becomes a multi-tiered process that extends far beyond initial fluorescence observation. While GFP expression serves as an excellent initial reporter for successful transfection or transduction [77], it does not confirm that the intended genetic alteration has occurred at the target locus. The GFP signal may indicate successful delivery of CRISPR constructs, but comprehensive validation requires confirmation at the DNA sequence level and, subsequently, at the functional protein level [76] [77]. This application note details a suite of validation techniques, from initial enzymatic screening to definitive sequencing methods, providing researchers with a structured framework to confidently confirm their CRISPR editing outcomes, particularly when working with GFP-marked transformants.
A robust CRISPR validation strategy employs a tiered approach, often beginning with rapid, accessible enzymatic assays for initial screening before progressing to more definitive, sequencing-based methods for precise characterization. This sequential methodology balances efficiency with analytical depth.
Initial Screening with Enzyme Mismatch Cleavage Assays
Enzyme mismatch cleavage (EMC) techniques, such as the T7 Endonuclease I (T7E1) assay, provide a cost-effective and rapid first-pass analysis for detecting CRISPR-induced mutations. These assays leverage enzymes that recognize and cleave DNA heteroduplexes formed when wild-type and mutant DNA strands reanneal [76] [78].
Protocol: T7 Endonuclease I (T7E1) Assay [76]
- Genomic DNA Isolation: Harvest cells and isolate genomic DNA from both CRISPR-edited and control cell populations.
- PCR Amplification: Amplify the genomic region surrounding the gRNA target site using a high-fidelity DNA polymerase (e.g., AccuTaq LA DNA Polymerase) to prevent PCR-introduced errors. Design primers to generate an amplicon of 500-1200 bp.
- DNA Denaturation and Reannealing: Purify the PCR products. Denature the DNA by heating (e.g., 95°C for 5-10 minutes) and then slowly cool to room temperature to allow strands to reanneal. This process creates heteroduplexes (wild-type/mutant paired strands) in addition to homoduplexes (wild-type/wild-type or mutant/mutant).
- T7E1 Digestion: Incubate the reannealed DNA with T7 Endonuclease I enzyme, which cleaves at the mismatch sites in the heteroduplex DNA.
- Analysis by Gel Electrophoresis: Run the digestion products on an agarose gel. Successful editing is indicated by the presence of additional, smaller DNA fragments resulting from the cleavage of heteroduplexes. Editing efficiency can be estimated by comparing the band intensity of cleaved versus uncleaved products using gel analysis software.
The T7E1 assay is a powerful initial screen but has limitations: it cannot determine the specific sequence of the induced mutations and may yield false positives from naturally occurring polymorphisms [76] [77].
Definitive Confirmation with Sequencing-Based Methods
Sequencing technologies provide the ultimate resolution for validating CRISPR edits, offering precise identification of insertion/deletion (indel) patterns and their frequencies within a cell population.
Sanger Sequencing and TIDE Analysis
Sanger sequencing is a reliable method for confirming edits in clonal cell populations. For mixed populations, the Tracking of Indels by Decomposition (TIDE) method provides a more efficient approach [76] [77].
Protocol: TIDE Analysis [76] [79]
- PCR and Sequencing: Amplify the target region from both edited and control (wild-type) cell populations using PCR. Purify the PCR products and perform Sanger sequencing for both samples.
- Data Analysis: Upload the Sanger sequencing chromatograms (from both edited and control samples) to the TIDE web tool (https://tide.nki.nl). The software algorithm decomposes the complex chromatogram from the edited sample by comparing it to the control reference, quantifying the spectrum and frequency of different indels.
- Interpretation: The TIDE output reports the types of indels present, their respective frequencies, and the overall editing efficiency, providing a quantitative assessment of the editing outcome without the need for subcloning.
Next-Generation Sequencing (NGS)
NGS offers the highest sensitivity and throughput for CRISPR validation, capable of detecting low-frequency mutations and profiling off-target effects across the genome [76] [80].
Protocol: Targeted Amplicon Sequencing for CRISPR Validation [80]
- Library Preparation (Two-Step PCR):
- First PCR: Amplify the target genomic site(s) from extracted gDNA using gene-specific primers that contain partial, overhanging Illumina adapter sequences.
- Second PCR: Use a second set of primers to attach full Illumina sequencing adapters, including unique sample indices (barcodes), to the amplicons from the first PCR. This allows multiple samples to be pooled and sequenced simultaneously.
- Sequencing: Pool the purified, barcoded libraries and sequence them on a high-throughput platform (e.g., Illumina MiSeq).
- Bioinformatic Analysis: Process the sequencing data through a specialized analysis tool such as CRISPResso. This software aligns the sequence reads to a reference amplicon sequence, precisely identifies and quantifies the induced indels, and generates a comprehensive report on editing efficiency and specificity.
Comparative Analysis of Validation Techniques
The choice of validation method depends on several factors, including the number of samples, required throughput, desired information depth, and available budget. The table below summarizes the key characteristics of the primary validation techniques.
Table 1: Comparison of Primary CRISPR Validation Methods
| Method | Key Principle | Throughput | Information Gained | Relative Cost | Key Applications |
|---|---|---|---|---|---|
| T7E1 Assay [76] [77] | Enzyme cleavage of DNA heteroduplexes | Low to Medium | Presence/Absence of indels; Estimated efficiency | $ | First-pass screening; Efficiency check |
| TIDE [76] [77] | Decomposition of Sanger sequencing traces | Low to Medium | Indel sequences and frequencies | $$ | Quick, cost-effective genotyping of mixed populations |
| NGS (Amplicon) [76] [80] [81] | Massive parallel sequencing of PCR amplicons | High | Comprehensive indel spectrum; Precise quantification; Off-target analysis | $$$ | Definitive validation; Sensitive detection of rare variants; Off-target assessment |
Integration with GFP-Based Visual Screening
In a research pipeline focused on visual screening of CRISPR transformants with GFP markers, these validation techniques integrate seamlessly into a comprehensive workflow. GFP fluorescence first confirms the delivery and expression of the CRISPR construct [77]. Flow cytometry can then be used to quantify transfection efficiency and sort the fluorescent cell population for further analysis [82] [79]. Following this enrichment, genomic DNA is extracted from the GFP-positive cells and subjected to the validation cascade—starting with a T7E1 assay for a quick efficiency check and culminating in NGS for definitive, quantitative genotyping [76] [80] [81].
It is critical to note that while GFP signals successful delivery, the functional loss of the target protein must be confirmed separately, typically by Western blotting using a validated antibody, as indels do not always guarantee complete protein knockout [76] [77].
Table 2: Essential Research Reagent Solutions for CRISPR Validation
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| High-Fidelity DNA Polymerase [76] | Accurate amplification of target locus for T7E1 or sequencing | Prevents false positives in T7E1 assay by minimizing PCR errors. |
| T7 Endonuclease I [76] [78] | Cleaves mismatched DNA heteroduplexes | Detection of indels in the T7E1 enzymatic cleavage assay. |
| Anti-GFP Antibodies [83] | Detect GFP fusion protein via flow cytometry or microscopy | Amplify GFP signal; enable channel switching to avoid autofluorescence. |
| NGS Library Prep Kits [78] | Prepare amplicon or whole-genome libraries for sequencing | Targeted sequencing of CRISPR edits using kits like NEBNext Ultra II. |
| CRISPR Analysis Software (e.g., TIDE, CRISPResso) [76] [80] | Bioinformatic analysis of sequencing data | Quantifies indel frequencies from Sanger or NGS data, respectively. |
Experimental Workflow and Visualization
The following diagram illustrates the integrated workflow for validating CRISPR edits, from initial visual screening to definitive genotyping.
A rigorous, multi-tiered validation strategy is indispensable for confirming successful genome editing in CRISPR research, especially when building upon initial visual screening with GFP markers. By combining the rapid screening capability of the T7E1 assay with the precise, quantitative power of sequencing-based methods like TIDE and NGS, researchers can confidently genotype their transformed cell populations. This structured approach from initial fluorescence observation to DNA-level confirmation ensures the reliability and interpretability of experimental results, forming a solid foundation for downstream functional analyses and therapeutic development.
Within the broader thesis on visually screening CRISPR transformants using GFP markers, a critical question emerges: how reliable is the fluorescent signal as a proxy for precise genetic edits? While GFP-based screening provides a rapid and high-throughput method for identifying potential transformants, its accuracy must be rigorously validated against sequencing data, the gold standard for confirming genomic alterations. This application note details protocols and quantitative assessments for comparing GFP-based results to sequencing data, providing researchers with a framework to evaluate the performance of their visual screening systems. The integration of these methods ensures that the convenience of fluorescent screening does not come at the cost of experimental accuracy, which is paramount in critical applications such as functional genomics and drug development.
Quantitative Comparison of GFP-Based Screening and Sequencing Outcomes
The table below summarizes key findings from studies that directly or indirectly compared GFP reporter results with sequencing-based validation methods.
Table 1: Comparison of GFP-Based Screening Outcomes with Sequencing Validation
| Study Context | GFP-Based Readout | Sequencing Validation Result | Correlation and Key Findings |
|---|---|---|---|
| HSV-1 Infection Tracking [84] | GFP expression from recombinant virus (GFP-McKrae) | PCR and Next-Generation Sequencing (NGS) | High correlation; 98% of infected cells were GFP+ and gD+ by flow cytometry; viral genome sequencing confirmed GFP insertion. |
| Plant CRISPR (pKSE401G Vector) [6] | GFP fluorescence in T1 seeds and seedlings | PCR amplicon sequencing (DSDecode analysis) | Effective correlation; GFP fluorescence successfully identified transformants, with sequencing revealing mutation frequencies from 20.4% to 90% across species. |
| HDR Reporter in Porcine Cells [65] | Fluorescence from a promoterless EGFP reporter knocked into GAPDH | N/A (Assessment of false positives) | Poor accuracy; high EGFP expression was detected even without Cas9/sgRNA, indicating the reporter is not exclusively expressed from HDR. |
Experimental Protocols for Cross-Validation
This section provides detailed methodologies for key experiments that utilize both GFP screening and sequencing to ensure accurate identification and isolation of genetically modified cells or organisms.
Protocol: Validation of a Fluorescent Reporter Virus Using Sequencing
This protocol, adapted from a study on HSV-1 pathogenesis, outlines the steps for creating and validating a recombinant virus expressing GFP [84].
- Application: To track viral infection and replication spatially and temporally, and to identify infected cell types, with confirmation that GFP expression faithfully represents the presence of the virus.
Key Reagents:
Procedure:
- Vector Construction: Engineer a recombinant virus (e.g., HSV-1 McKrae strain) to express GFP under a strong constitutive promoter (e.g., CMV). Insert the GFP gene into a genomic region that does not disrupt essential viral genes, such as the space between gJ and gD [84].
- In Vitro Characterization:
- Infect susceptible cells (e.g., RS cells) with the GFP-reporter virus.
- Confirm GFP expression via confocal microscopy and flow cytometry.
- Co-stain cells with an antibody against a viral protein (e.g., anti-gD) and analyze by flow cytometry to determine the percentage of cells that are both GFP+ and gD+ [84].
- Molecular Validation:
- Extract viral DNA from purified virions or infected cells.
- Perform PCR across the GFP insertion site and sequence the amplicons to confirm correct integration.
- Subject the entire viral genome to Next-Generation Sequencing (NGS) to verify the GFP sequence and ensure no other unintended mutations are present [84].
- Functional Assays: Compare the replication kinetics and pathogenicity of the GFP-reporter virus to the wild-type parent virus using standard plaque assays in vitro and in animal models to ensure the reporter does not attenuate the virus [84].
Protocol: Isolation of Transgene-Free CRISPR Mutants in Plants Using GFP
This protocol uses a GFP visual marker to streamline the identification of positive primary transformants and, crucially, the isolation of transgene-free edited plants in subsequent generations [6].
- Application: Rapid screening of CRISPR/Cas9 transformants and efficient isolation of non-transgenic mutant lines in Arabidopsis, B. napus, strawberry, and soybean.
Key Reagents:
- pKSE401G CRISPR/Cas9 Vector: A modified vector containing a 35S::sGFP cassette for visual screening [6].
Procedure:
- Vector Design and Transformation:
- Clone two sgRNAs targeting your gene of interest into the pKSE401G vector using Golden Gate assembly.
- Transform the construct into your plant species of choice using Agrobacterium-mediated transformation or other suitable methods [6].
- T1 Generation (Primary Transformants):
- Screen seeds or young seedlings for GFP fluorescence using a stereo fluorescence microscope. GFP-positive individuals are potential transgenic mutants.
- From GFP-positive plants, extract genomic DNA and amplify the target region by PCR.
- Sequence the PCR products using Sanger sequencing and decode the sequencing chromatograms using a tool like DSDecode to determine the exact mutation types and frequencies [6].
- T2/T1 Generation (Isolation of Transgene-Free Mutants):
- Harvest seeds from confirmed mutant T1 plants.
- Screen the next generation (T2 for Arabidopsis, T1 for B. napus) for the absence of GFP fluorescence.
- These GFP-negative plants are potential transgene-free mutants. Genotype these plants by sequencing the target locus to confirm they have retained the desired mutation but lost the CRISPR/Cas9-GFP T-DNA [6].
- Stability Check: Ensure the mutations are stably inherited by sequencing the progeny of the transgene-free mutants.
- Vector Design and Transformation:
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and their functions for conducting and validating GFP-based CRISPR screening experiments.
Table 2: Essential Reagents for GFP-Based CRISPR Screening and Validation
| Reagent | Function/Application | Examples/Notes |
|---|---|---|
| GFP Reporter Vectors | Serve as visual markers for transfection/transformation and CRISPR activity. | pKSE401G for plants [6]; GFP-McKrae for viral tracking [84]. |
| Anti-GFP Antibodies | Detect GFP via flow cytometry or immunofluorescence; amplify signal, overcome GFP fluorescence issues (e.g., from fixation). | Rabbit Polyclonal Anti-GFP; Alpaca VHH Anti-GFP fragments [83]. |
| NGS Services/Kits | Validate CRISPR edits genome-wide; detect off-target effects and complex structural variations. | Used for whole viral genome validation [84] and transcriptome analysis in CRISPR KO lines [85]. |
| sgRNA Cloning Vector | Platform for sgRNA assembly and expression. | U6-sgRNA vector for cloning annealed oligonucleotides [65]. |
| Flow Cytometry Reagents | Quantify GFP-positive cell populations and sort them for further expansion or analysis. | Staining buffers (PBS + 0.2% BSA) [83]. |
Workflow Diagrams for Validation Strategies
The following diagram illustrates the logical workflow for validating GFP-based screening results against sequencing data, integrating the protocols described above.
Validating GFP Reports with Sequencing
Critical Considerations and Limitations
While GFP screening is powerful, awareness of its limitations is crucial for accurate data interpretation.
- Risk of False Positives: Promoterless GFP reporters designed to only express upon correct homologous recombination can sometimes exhibit high background expression without the intended genetic event, leading to false positives [65]. Sequencing validation remains essential.
- Fluorescence Limitations: GFP fluorescence can be compromised by low pH, anaerobic conditions, or sample fixation processes. In such cases, using anti-GFP antibodies for detection can rescue the signal, providing greater experimental flexibility [83].
- Beyond DNA Sequencing: CRISPR/Cas9 can induce complex, unanticipated transcriptional changes, such as exon skipping, gene fusions, and large deletions, that are not detectable by PCR amplification of the DNA target site alone. RNA-seq provides a more comprehensive validation of the transcriptional outcome of a gene knockout [85].
Within the broader context of visual screening for CRISPR transformants, GFP markers serve as a powerful initial readout for successful gene editing. However, comprehensive validation requires a multi-faceted approach that extends beyond fluorescence. This application note details a rigorous framework for verifying CRISPR outcomes, integrating rapid GFP-based enrichment with definitive protein-level (Western Blot) and physiological (Functional Assays) confirmation. This multi-method strategy is crucial for generating high-confidence data in research and drug development, ensuring that observed phenotypic changes are directly linked to the intended genomic and proteomic alterations.
The core advantage of this workflow lies in its hierarchical verification logic. GFP screening provides a rapid, high-throughput, and visual means to identify potential positive transformants. Subsequent Western Blot analysis confirms the presence, size, and relative abundance of the protein product resulting from the edit. Finally, functional assays determine whether the edited protein is physiologically active, bridging the gap between molecular modification and biological outcome. This cascading confirmation is especially critical when working with hypomorphic alleles or when the GFP readout is indirect, as it mitigates the risk of false positives and provides a complete picture of the editing event's consequences.
The Essential Workflow for Multi-Method Verification
The following diagram illustrates the integrated experimental pathway from initial cell preparation to final validated clone selection.
Key Research Reagent Solutions
Successful execution of the verification workflow depends on critical reagents and tools. The table below catalogues essential solutions for implementing this multi-method approach.
Table 1: Key Research Reagents and Tools for CRISPR Verification Workflows
| Reagent/Tool | Primary Function | Application Context |
|---|---|---|
| GFP-on Reporter Mouse Model [40] | In vivo reporter for adenine base editing (ABE) efficiency; contains a correctable nonsense mutation in EGFP. | Validating base editor delivery and activity across tissues in adult and fetal mice. |
| Programmable Gene Editors (e.g., OpenCRISPR-1) [86] | AI-designed Cas9 effector for precision genome editing with high activity and specificity. | Core editing tool for introducing genetic modifications in various cell types. |
| Leo System for Simple Western [87] | Fully automated, high-throughput Western blotting; enables absolute protein quantification. | Potency testing for AAV vectors; sensitive quantification of transgene protein expression (e.g., GFP). |
| Adeno-Associated Virus (AAV9) [40] | In vivo delivery vehicle for CRISPR components; offers tunable tropism for targeted cell types. | Systemic delivery of base editors and sgRNAs in animal models. |
| Ribonucleoproteins (RNPs) [88] | Pre-complexed Cas9 protein and guide RNA for direct delivery into cells. | Reduces off-target effects; enables rapid editing, especially in sensitive primary cells. |
| Auxin-Inducible Degron (AID2) System [89] | Targeted, rapid degradation of GFP-tagged proteins of interest. | Functional validation by creating acute hypomorphic conditions, mimicking disease states. |
Experimental Protocols & Data Analysis
Protocol: GFP-Based Screening and Enrichment
This protocol begins after the delivery of CRISPR components into your target cells.
- Cell Culture and Transfection: Culture cells appropriate for your experiment (e.g., immortalized cell lines, primary cells, or stem cells). Select and execute a transfection protocol optimized for your cell type. For sensitive primary cells, nucleofection of RNP complexes is highly effective, while lipofection may suffice for standard immortalized lines [88].
- Incubation and Expression: Allow cells to recover and express the edited genome for 48-72 hours. This period allows for the turnover of existing proteins and the expression of the GFP reporter.
- Harvesting: Gently dissociate adherent cells using a non-enzymatic cell dissociation solution or trypsin-EDTA to preserve cell surface integrity and GFP fluorescence.
- Flow Cytometry Analysis and Sorting:
- Resuspend the cell pellet in a cold, protein-based FACS buffer (e.g., PBS with 1-2% FBS) to maintain cell viability.
- Pass the cell suspension through a flow cytometer equipped with a 488 nm laser for excitation and a 530/30 nm bandpass filter for GFP detection.
- Establish gating parameters using non-transfected control cells to define the background fluorescence and set the threshold for GFP positivity.
- Sort the population of GFP-positive cells into a collection tube containing growth media. This enriched pool is used for downstream verification steps.
Protocol: Western Blot Verification
This protocol confirms the presence and molecular weight of the GFP-tagged or edited protein.
- Protein Lysate Preparation: Lyse the sorted GFP-positive cells and a negative control cell sample in RIPA buffer supplemented with protease inhibitors. Incubate on ice for 30 minutes, followed by centrifugation at 14,000 x g for 15 minutes to clear the lysate.
- Gel Electrophoresis and Transfer:
- Mix equal amounts of total protein (20-30 µg) with Laemmli sample buffer, denature at 95°C for 5 minutes, and load onto a 4-20% gradient SDS-polyacrylamide gel.
- Separate proteins by electrophoresis at constant voltage.
- Transfer proteins from the gel to a PVDF or nitrocellulose membrane using a wet or semi-dry transfer system.
- Immunoblotting:
- Block the membrane with 5% non-fat milk in TBST for 1 hour at room temperature.
- Incubate with a primary antibody against your protein of interest (if not GFP) and/or an anti-GFP antibody (e.g., 1:1000 dilution) in blocking buffer overnight at 4°C.
- Wash the membrane 3 times for 5 minutes each with TBST.
- Incubate with an appropriate HRP-conjugated secondary antibody (1:2000-5000 dilution) for 1 hour at room temperature.
- Wash again as before.
- Detection and Analysis: Develop the blot using a chemiluminescent substrate and image with a digital imager. The presence of a single band at the expected molecular weight confirms successful editing and protein expression. For high-throughput, quantitative applications, automated systems like the Leo System can be employed for absolute protein quantification with minimal hands-on time [87].
Protocol: Functional Assay for Edited Proteins
The nature of this assay depends entirely on the protein's function. The example below is for an enzyme, but it should be adapted accordingly.
- Sample Preparation: Prepare lysates from the verified GFP-positive/Western-positive cells and from control (non-edited or wild-type) cells in a non-denaturing lysis buffer.
- Reaction Setup:
- Combine cell lysate (containing the enzyme) with the specific substrate for your protein in an appropriate reaction buffer.
- For a PMM2 enzyme assay, as used in a medaka fish model, the reaction would involve monitoring the conversion of mannose-6-phosphate to mannose-1-phosphate [89].
- Include a no-enzyme control (substrate only) to account for non-specific substrate turnover.
- Incubation and Measurement: Incubate the reaction at the enzyme's optimal temperature (e.g., 37°C) and measure the product formation over time using a method specific to the reaction (e.g., spectrophotometry, fluorescence, mass spectrometry).
- Data Analysis: Calculate the enzyme's specific activity (e.g., units per mg of total protein) for the edited and control samples. A significant change in activity in the edited sample confirms a functional outcome from the CRISPR edit.
Data Integration and Interpretation
The verification cascade culminates in the integration of data from all three methods. The following diagram illustrates the logical relationship and how evidence from each level builds toward a definitive conclusion.
Quantitative data from these assays should be compiled for a clear comparison. The table below provides a template for organizing key results from the verification process.
Table 2: Template for Compiling Multi-Method Verification Data
| Cell Line / Sample ID | GFP Positivity (%) | Western Blot Result (Band Size) | Functional Assay Result | Final Verification Status |
|---|---|---|---|---|
| HEK293 (Control) | < 0.5% | No band at target size | Baseline Activity (100%) | Negative |
| HEK293 (Edited Pool) | 45.5% | Single band at ~55 kDa | 215% of Control | Positive - Gain of Function |
| HSC (Edited Pool) | 18.2% | Single band at ~27 kDa | 15% of Control | Positive - Hypomorphic |
This integrated approach is exemplified by research using a GFP-on mouse model, where GFP restoration was first visualized, then quantified by flow cytometry, and ultimately linked to the successful in vivo delivery and activity of adenine base editors [40]. Similarly, the functional consequence of protein knockdown, achieved via a GFP-nanobody degron system, was confirmed not just by loss of fluorescence but also by a direct enzymatic assay showing reduced activity to pathogenic levels [89].
In the field of CRISPR-based genome editing, the accurate measurement of on-target editing efficiency is crucial for developing robust research and therapeutic applications. While visual screening of transformants using GFP markers provides rapid functional readouts, researchers often rely on biochemical assays for definitive quantification of editing events. Among these, the T7 Endonuclease I (T7EI) assay has persisted as a widely used method due to its procedural simplicity and minimal instrumentation requirements. However, a growing body of evidence indicates that this assay can significantly misrepresent true editing efficiency, potentially leading to flawed experimental conclusions. This application note examines the specific limitations of the T7EI assay and presents superior alternative methods, with particular emphasis on quantitative approaches that align with visual screening methodologies using fluorescent reporters. Understanding these limitations is especially critical for researchers employing GFP-based systems, as discrepancies between biochemical and functional readouts can complicate the interpretation of editing outcomes.
Understanding T7EI Assay Limitations
The T7 Endonuclease I assay operates on the principle of mismatch cleavage, where the T7EI enzyme recognizes and cleaves heteroduplex DNA formed by annealing wild-type and edited DNA strands. The cleavage products are separated by gel electrophoresis, and editing efficiency is estimated semi-quantitatively through densitometric analysis of band intensities [90]. While straightforward to implement, this methodology contains several fundamental limitations that impact data accuracy.
Semi-Quantitative Nature: The T7EI assay provides only approximate efficiency measurements through band intensity ratios, lacking the precision of fully quantitative methods. Densitometric analysis of agarose gels introduces substantial variability, with typical error margins of 10-15% even under optimized conditions [90].
Sensitivity Constraints: The assay demonstrates limited sensitivity, particularly for detecting low-frequency editing events. The minimum detection threshold typically ranges between 2-5%, rendering it ineffective for evaluating modest editing efficiencies often encountered in primary cells or challenging target loci [90].
Sequence Context Dependence: Cleavage efficiency by T7EI varies significantly based on the specific mismatch configuration and surrounding sequence context. This variability can lead to substantial under- or over-estimation of true editing frequencies depending on the induced mutation [90].
Inability to Characterize Mutation Profiles: While the assay indicates the presence of mutations, it provides no information about the specific types of edits induced (insertions, deletions, or base substitutions), which is crucial for understanding functional outcomes [90].
Table 1: Key Limitations of T7 Endonuclease I Assay
| Limitation | Impact on Data Accuracy | Experimental Consequence |
|---|---|---|
| Semi-quantitative readout | High variance in efficiency estimates | Poor reproducibility between experiments |
| Low sensitivity | Failure to detect low-frequency editing events | Overestimation of editing specificity |
| Sequence-dependent cleavage efficiency | Inconsistent correlation between cleavage and actual editing | Under/over-estimation of true efficiency |
| No sequence information | Unknown mutation profiles | Limited insight into functional outcomes |
Comparative Analysis of Editing Efficiency Assessment Methods
Recent methodological comparisons reveal significant performance differences between T7EI and alternative editing assessment platforms. These findings demonstrate that the choice of detection method substantially impacts the perceived success of genome editing experiments.
Quantitative Performance Metrics
Advanced comparative studies utilizing controlled plasmid mixtures with predefined editing ratios have quantified the performance disparities between methods. When analyzing samples with known editing efficiencies ranging from 5% to 95%, T7EI consistently demonstrated the highest variance and poorest correlation with expected values (R² = 0.89), while droplet digital PCR (ddPCR) achieved near-perfect correlation (R² = 0.99) [90]. Sanger sequencing-based methods like TIDE and ICE showed intermediate performance (R² = 0.93-0.96), representing a substantial improvement over T7EI while remaining accessible to most molecular laboratories.
Sensitivity and Dynamic Range
The critical lower limit of detection varies considerably between methods. While T7EI struggles to reliably detect editing below 5%, TIDE and ICE can accurately quantify efficiencies as low as 1-2% [90]. Digital PCR methods offer exceptional sensitivity, detecting edits at frequencies of 0.1% or lower, making them particularly valuable for assessing editing in heterogenous cell populations [90]. Fluorescent reporter systems provide an intermediate sensitivity level but offer the unique advantage of enabling live-cell enrichment and tracking of edited cells, bridging the gap between biochemical confirmation and functional assessment [91].
Table 2: Performance Comparison of CRISPR Editing Efficiency Assessment Methods
| Method | Sensitivity | Quantitative Capability | Mutation Characterization | Throughput |
|---|---|---|---|---|
| T7 Endonuclease I | ~5% | Semi-quantitative | No | Medium |
| TIDE/ICE | ~1-2% | Quantitative | Yes (indels) | Medium |
| ddPCR | ~0.1% | Highly quantitative | Limited (predesigned) | High |
| Fluorescent Reporters | ~1% * | Semi-quantitative * | No (but functional readout) | High |
| Next-Generation Sequencing | ~0.01% | Quantitative | Comprehensive | Low-Medium |
Sensitivity for fluorescent reporters depends on flow cytometry detection thresholds and reporter design.
Fluorescent Reporter Systems: Bridging Quantitative Assessment and Functional Screening
Engineered fluorescent reporter systems provide a powerful alternative for assessing CRISPR editing efficiency while simultaneously enabling visual screening and enrichment of successfully edited cells. These systems bridge the gap between biochemical confirmation and functional assessment, offering unique advantages for tracking editing outcomes in live cells.
GFP Activation Assay Principle and Applications
The GFP activation assay employs a clever design where a target sequence is inserted between the start codon and the GFP coding sequence, disrupting GFP expression through a frameshift. Successful CRISPR-mediated cleavage and subsequent non-homologous end joining (NHEJ) repair can restore the reading frame in a subset of cells, leading to GFP expression that can be quantified by flow cytometry or fluorescence microscopy [91]. This system provides direct functional readouts of editing efficiency while enabling live-cell tracking and enrichment of edited populations.
The exceptional sensitivity of GFP activation assays has been demonstrated in applications detecting rare off-target cleavage events that were undetectable by conventional targeted amplicon sequencing. In one study, GFP reporters confirmed off-target cleavage at sites previously identified only by CIRCLE-seq but not validated in cells by sequencing methods, highlighting their utility for comprehensive specificity profiling [91].
Dual-Fluorescent Reporter Systems for Enrichment
Advanced reporter designs incorporate multiple fluorescent markers to enable both efficiency assessment and cell enrichment. The RFP-GFP-GFP reporter system constitutively expresses mRFP regardless of editing, serving as a transfection control and normalization marker, while two out-of-frame GFP genes remain silent until CRISPR-induced indels restore the reading frame [92]. This design permits precise quantification of editing efficiency as the ratio of GFP+/RFP+ cells while enabling fluorescence-activated cell sorting (FACS) to isolate edited populations for downstream applications.
Diagram 1: Dual-Fluorescent Reporter Workflow for CRISPR Efficiency Assessment and Cell Enrichment
Experimental Protocols for Superior Efficiency Assessment
GFP Activation Assay for Sensitive Off-Target Detection
Protocol Overview: This method enables highly sensitive detection of DNA cleavage events in cells through frame restoration of GFP.
Materials:
- HEK293T cells or other relevant cell line
- GFP reporter construct (lentiviral backbone)
- SpCas9 expression plasmid
- Target-specific gRNA expression plasmid
- Flow cytometer or fluorescence microscope
- Cell sorting capability (optional)
Procedure:
- Generate stable reporter cell line by lentiviral delivery of GFP reporter construct
- Remove background GFP-positive cells by FACS to establish clean baseline
- Transfect with SpCas9 and target-specific gRNA (test and control groups)
- Analyze GFP expression by flow cytometry 48-72 hours post-transfection
- Sort GFP-positive populations for molecular validation if required
- Amplify and sequence target sites from sorted populations to confirm indels
Validation: This protocol has been successfully employed to verify rare off-target cleavage events that could not be detected by targeted amplicon sequencing, demonstrating superior sensitivity for comprehensive CRISPR specificity profiling [91].
Stable Dual-Fluorescent Reporter Cell Line Generation
Protocol Overview: Establishment of stable cell lines expressing the RFP-GFP-GFP reporter for CRISPR efficiency quantification and enrichment.
Materials:
- pRG2S_Cas9 reporter plasmid or equivalent
- HEK293 cells or other suitable cell line
- G418 antibiotic for selection
- Restriction enzymes (BstXI and BamHI)
- FuGENE transfection reagent or equivalent
- Flow cytometer with sorting capability
Procedure:
- Clone desired Cas9 target sequences into reporter plasmid using BstXI and BamHI sites
- Transfect cells with reporter construct using appropriate transfection method
- Select stable integrants with G418 antibiotic (typically 1-2 weeks)
- Sort RFP-positive population by FACS to establish pure reporter line
- Validate reporter function with control gRNAs with known activity
- Use established line for evaluating novel gRNAs or optimizing delivery methods
Applications: This system enables real-time quantification of nuclease activity and allows enrichment of edited cells, significantly accelerating the generation of knockout cell lines [92].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for CRISPR Efficiency Assessment
| Reagent/Category | Function | Application Notes |
|---|---|---|
| T7 Endonuclease I | Mismatch-specific cleavage | Use limited to semi-quantitative assessment; requires PCR amplification |
| ICE Analysis Software | Decomposition of Sanger sequencing traces | Free web tool; provides quantitative efficiency and mutation profiles |
| GFP Activation Reporter | Frame-based editing detection | Enables live-cell tracking and rare event detection |
| Dual RFP-GFP Reporter | Efficiency quantification & enrichment | Integrated control for transfection and editing assessment |
| ddPCR Mutation Detection | Absolute quantification of edits | Requires specific probe design; exceptional sensitivity and precision |
| Positive Control gRNAs | Experimental validation | Target conserved genes (e.g., TRAC, RELA) with known high efficiency |
| Scramble gRNA | Negative control | No genomic target; establishes baseline for non-specific effects |
The limitations of the T7 Endonuclease I assay present significant challenges for accurate quantification of CRISPR editing efficiency, particularly when correlated with functional outcomes in visual screening systems. While T7EI may suffice for preliminary assessment in applications where approximate efficiency is acceptable, researchers requiring precise quantification should implement more advanced methods. For most applications requiring balance between practicality and precision, TIDE/ICE analysis of Sanger sequencing data represents a substantial improvement over T7EI while remaining broadly accessible. When maximum sensitivity and absolute quantification are essential, ddPCR provides exceptional performance. Fluorescent reporter systems offer the unique advantage of linking biochemical confirmation with functional assessment in live cells, enabling both efficiency measurement and enrichment of edited populations. The integration of these validated methods with appropriate positive and negative controls will significantly enhance the reliability and reproducibility of CRISPR editing assessments, particularly in the context of visual screening workflows employing GFP and other fluorescent markers.
Alternative Reporter Systems: Comparing GFP with Luciferase and Other Markers
The selection and analysis of genetically modified cells is a cornerstone of modern biological research, particularly in the field of CRISPR-based genome editing. Reporter systems, which allow scientists to track gene expression, protein localization, and cellular events, are indispensable tools in this process. Among the most prominent are Green Fluorescent Protein (GFP) and luciferase, each with distinct mechanisms and applications. A recent, direct comparative study demonstrates a clear superiority of GFP for in vivo imaging applications, showing stronger, more stable signals and a 300-fold faster detection time compared to luciferase-luciferin systems [93]. This application note provides a detailed comparison of these and other reporter systems, framed within the context of visual screening of CRISPR transformants, to guide researchers in selecting the optimal tool for their experimental needs.
Head-to-Head Comparison: Quantitative Data
The following table summarizes key performance metrics from a direct comparative study of GFP and luciferase, providing a quantitative basis for system selection.
Table 1: Quantitative Comparison of GFP and Luciferase Reporter Systems In Vivo
| Performance Metric | GFP Fluorescence | Luciferase-Luciferin |
|---|---|---|
| Signal Intensity at 10 min | 56,186 (arbitrary units) | 28,065 (arbitrary units) |
| Signal Intensity at 20 min | 57,085 (maintained) | 5,199 (~80% decrease) |
| Signal Stability | High (maintained over 20 min) | Low (rapidly bioluminescent decay) |
| Required Exposure Time | 100 milliseconds | 30 seconds |
| Excitation/Emission | Excitation: 487 nm, Emission: 513 nm | Emission: 560 nm |
Data adapted from Mizuta et al. 2024 [93]
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and their functions for implementing reporter systems, particularly in the context of CRISPR screening workflows.
Table 2: Research Reagent Solutions for Reporter-Based CRISPR Screening
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| sgRNA Library [94] [95] [96] | A pooled collection of single guide RNAs targeting multiple genes for large-scale functional genomics screens. | Libraries can be genome-wide (e.g., GeCKO, Brunello) or targeted. Design includes multiple sgRNAs per gene and non-targeting controls. |
| Lentiviral Vectors [95] [96] | Delivery of CRISPR components (e.g., Cas9, sgRNAs) or reporter constructs into host cells, including primary and hard-to-transfect cells. | Enables stable genomic integration. Low multiplicity of infection (MOI) ensures single sgRNA integration per cell. |
| Stable Reporter Cell Lines [61] | Engineered cells with a reporter construct (e.g., RFP-GFP) integrated into the genome for quantifying CRISPR nuclease activity and transfection efficiency. | Recapitulates endogenous gene targeting. Allows for high-throughput, microplate-reader based quantification of editing efficiency. |
| Dual-Fluorescence Reporter (e.g., RFP-GFP) [61] | A construct used to detect and enrich cells with successful CRISPR-Cas9-induced mutations. | RFP constitutively expresses for normalization. GFP activates only upon successful NHEJ repair, signaling functional gene editing. |
| Cas9-Expressing Cell Lines [96] | Host cells that stably express the Cas9 nuclease, simplifying the screening process by requiring only sgRNA delivery. | Provides uniform Cas9 expression, improving editing consistency and efficiency across the cell population. |
| Lipid-Based Transfection Reagents [61] | Delivery of CRISPR components, such as Cas9/sgRNA ribonucleoprotein (RNP) complexes, into cells. | Efficiency varies by cell type. Requires optimization to balance high delivery efficiency with low cytotoxicity. |
Experimental Protocol: Rapid Assessment of CRISPR Editing Efficiency with a Dual-Fluorescence Reporter
This protocol details the use of a stable dual-fluorescence (RFP-GFP) reporter cell line for the rapid, high-throughput quantification of CRISPR-Cas9 nuclease activity, a critical step in validating transfection methods and enriching for edited cells [61].
Principle
The stable reporter cell line constitutively expresses mRFP. A stop codon, flanked by a CRISPR target sequence, is placed between the RFP and out-of-frame eGFP genes. Successful CRISPR-Cas9 cutting at the target site triggers error-prone Non-Homologous End Joining (NHEJ) repair. The resulting insertions or deletions (indels) can bypass the stop codon, shifting one of the two eGFP copies into frame and resulting in permanent eGFP expression. The percentage of double-positive (RFP+GFP+) cells directly corresponds to the functional uptake and activity of the CRISPR machinery [61].
Materials
- Stable dual-fluorescence reporter cell line (e.g., HEK293-pRG2S_Cas9) [61]
- Functional CRISPR-Cas9 components (e.g., plasmid expressing Cas9 and sgRNA, or pre-formed RNP complexes)
- Appropriate cell culture medium and reagents
- Transfection reagent (e.g., lipofection agents)
- Multi-channel micropipettes
- Fluorescence microplate reader (e.g., FLUOstar Omega) capable of detecting RFP and GFP
- (Optional) Flow cytometer for validation
Procedure
Day 1: Cell Seeding
- Harvest the stable reporter cells and resuspend in complete growth medium.
- Seed cells into a 96-well microplate at a defined, optimized density (e.g., 10,000 cells per well in 100 µL of medium). Ensure multiple wells are reserved for control groups (e.g., non-transfected cells, cells transfected with a non-targeting sgRNA).
- Incubate the plate overnight at 37°C and 5% CO₂.
Day 2: Transfection
- Prepare the CRISPR-Cas9 delivery complexes according to the manufacturer's instructions for your chosen transfection reagent. This could be plasmid DNA or pre-formed RNP complexes targeting the sequence in the reporter construct.
- Add the complexes to the assigned wells. Include appropriate controls.
- Gently mix the plate and return it to the incubator for 48-72 hours to allow for gene editing and GFP expression.
Day 4/5: Microplate Reader Analysis
- Following incubation, ensure cells are healthy and confluent.
- Using the microplate reader, measure the fluorescence intensity for both RFP and GFP for each well, using appropriate excitation/emission filters (e.g., ~587/610 nm for RFP; ~488/510 nm for GFP).
- Calculate the GFP/RFP fluorescence intensity ratio for each test well.
- Using a pre-established standard curve (generated by plotting the GFP/RFP ratio against known percentages of RFP+GFP+ cells counted via flow cytometry), determine the percentage of nuclease-active (RFP+GFP+) cells in your population [61].
Data Analysis
- A higher percentage of GFP-positive cells indicates more efficient delivery and functional activity of the CRISPR-Cas9 system.
- Compare the editing efficiency across different transfection reagents, CRISPR delivery formats (plasmid vs. RNP), or sgRNA designs.
- This rapid assay allows for the high-throughput optimization of transfection conditions before applying them to more valuable, hard-to-obtain primary cells or complex model systems.
Visualizing the CRISPR Screening Workflow with Reporter Systems
The following diagram illustrates a generalized workflow for a pooled CRISPR knockout screen, a common application where reporter systems are employed for validation and analysis.
CRISPR Screening and Validation Workflow
The choice between GFP, luciferase, and other reporter systems is not one-size-fits-all. For applications requiring rapid, stable, and high-resolution spatial imaging—such as the initial visual screening and enrichment of CRISPR transformants—GFP-based systems offer significant advantages, as quantified by recent direct comparisons [93]. The development of advanced tools, such as stable dual-fluorescence reporter cell lines and sensitive detection systems, further enhances the utility of fluorescent proteins in quantitative, high-throughput workflows [61]. By carefully matching the strengths of each reporter technology to their experimental goals, researchers can optimize the efficiency and reliability of their CRISPR screening and functional genomics studies.
In the field of functional genomics, CRISPR-based screening has emerged as a powerful, high-throughput method for identifying genes that influence specific cellular phenotypes. The integration of GFP markers as visual reporters enables researchers to track transfection efficiency, monitor CRISPR activity, and sort cells based on phenotypic responses in screens designed to uncover genetic modifiers of drug sensitivity, essential genes, or other biologically relevant processes [12] [97]. However, the initial identification of candidate genes from a primary screen represents merely the starting point of discovery. The transition from a list of potential hits to biologically validated, high-confidence targets demands a rigorous, multi-stage validation strategy. False positives can arise from various sources, including off-target effects, sgRNA-specific artifacts, and technical variability inherent to high-throughput methodologies [98] [97]. This application note details a comprehensive framework of best practices for interpreting and validating screening hits, with a specific focus on workflows incorporating visual markers like GFP to enhance reliability and reproducibility.
Hit Identification from Primary Screens
The first step in the validation pipeline is the accurate interpretation of data from the primary screen. Pooled CRISPR screens, whether using knockout (CRISPRko), interference (CRISPRi), or activation (CRISPRa) systems, generate complex datasets where the abundance of each single-guide RNA (sgRNA) is quantified under selective pressure versus a reference baseline [98] [97].
Data Analysis and Hit Calling
Bioinformatics tools are essential for processing next-generation sequencing data, normalizing read counts, and identifying significantly enriched or depleted sgRNAs. Key analytical considerations include:
- Normalization: Adjusting for variations in library size and sequencing depth between samples.
- sgRNA-Level Analysis: Statistical testing (e.g., using negative binomial models) to identify sgRNAs with significant abundance changes.
- Gene-Level Analysis: Aggregating the effects of multiple sgRNAs targeting the same gene to improve confidence. Common algorithms for this include Robust Rank Aggregation (RRA) and Maximum Likelihood Estimation (MLE) [98].
The table below summarizes prominent computational tools for analyzing different types of CRISPR screens.
Table 1: Bioinformatics Tools for Analyzing CRISPR Screen Data
| Tool Name | Primary Application | Statistical Methodology | Key Features |
|---|---|---|---|
| MAGeCK [98] | Knockout Screens | Negative Binomial + RRA | Widely used; identifies positive and negative selection |
| MAGeCK-VISPR [98] | Integrated Workflow | Negative Binomial + MLE | Comprehensive pipeline with quality control |
| BAGEL [98] | Knockout Screens | Bayesian Factor | Uses a reference set of essential/non-essential genes |
| DrugZ [98] | Chemogenetic Screens | Normal Distribution + Z-score | Specifically for gene-drug interactions |
| CRISPhieRmix [98] | Diverse Screen Types | Hierarchical Mixture Model | Handles data from multiple screen modalities |
Hits are typically defined as genes whose targeting sgRNAs demonstrate a statistically significant and consistent fold-change in abundance, with a false discovery rate (FDR) below a predetermined threshold (e.g., 5%). For a GFP-based viability screen, "hits" would be genes whose knockout causes a significant change in the GFP-positive or GFP-negative cell population after selection.
Computational and Comparative Validation
Once a candidate list is generated, computational checks and cross-referencing with public databases provide a rapid, initial layer of validation, helping to prioritize candidates for downstream experimental work.
Interrogation of the Open Repository of CRISPR Screens (ORCS)
Resources like the BioGRID Open Repository of CRISPR Screens (ORCS) are invaluable for cross-validation [99]. This database houses over 890 manually curated CRISPR screens from published studies. Researchers can query their candidate genes against this resource to determine:
- If the gene has been identified as a hit in other published screens with similar or related phenotypes.
- The consistency of the gene's effect across different cellular models, libraries, and selection conditions. This independent confirmation significantly bolsters the credibility of a candidate hit.
Experimental Validation of Screening Hits
While computational validation is informative, experimental confirmation is indispensable. A multi-faceted approach is required to rule out false positives and confirm the biological role of the candidate gene.
Validation with Individual sgRNAs
The most critical step is to test whether the phenotype observed in the pooled screen can be recapitulated using individual sgRNAs outside the library context [12]. The recommended protocol is as follows:
- Protocol: Isolated sgRNA Validation
- Cloning: Clone at least 3-4 independent sgRNAs (including the top sgRNAs from the primary screen) targeting the candidate gene into a lentiviral transfer plasmid.
- Lentiviral Production: Produce lentivirus for each individual sgRNA separately.
- Cell Transduction: Transduce the Cas9-expressing cell line at a low MOI to ensure single-copy integration. A GFP reporter can be included in the vector to facilitate tracking of transduction efficiency and sorting of infected cells [12].
- Phenotypic Assay: Subject the transduced cells to the same selective conditions used in the primary screen.
- Analysis: Monitor the phenotype (e.g., cell viability via ATP assay, GFP intensity via flow cytometry, or imaging). Compare to cells transduced with non-targeting control (NTC) sgRNAs. A valid hit will show a consistent phenotype across multiple independent sgRNAs.
Diagram: The workflow for validating individual sgRNA hits.
Employing Alternative CRISPR Modalities
To minimize the risk of sgRNA-specific or mechanistic artifacts, employ orthogonal CRISPR tools. If the primary screen was performed with CRISPRko, validate key hits using CRISPR interference (CRISPRi) for transcriptional repression [12] [98]. The use of a different mechanism to perturb the same gene (transcriptional repression vs. gene knockout) that results in a concordant phenotype provides powerful confirmation of the gene's role. The following protocol outlines the establishment of a CRISPRi validation system:
- Protocol: Validation using an Inducible CRISPRi System
- Cell Line Engineering: Generate a stable cell line expressing a doxycycline-inducible dCas9-KRAB fusion protein and a reporter (e.g., mCherry) [12].
- sgRNA Design: Design sgRNAs to target the promoter region of the candidate gene.
- Transduction and Induction: Transduce cells with lentivirus delivering the gene-specific sgRNA, then induce dCas9-KRAB expression with doxycycline.
- Phenotypic Confirmation: Measure the phenotypic outcome (e.g., loss of GFP signal under selective pressure) and confirm knockdown of the target gene at the mRNA or protein level.
Orthogonal Functional Assays
Finally, the functional impact of the candidate gene should be confirmed using non-CRISPR methods. This provides the highest level of confidence that the observed phenotype is due to the loss of the target gene and not an artifact of the CRISPR system.
- Rescue Experiments: Re-introduce a CRISPR-resistant, wild-type cDNA of the candidate gene into the knockout cells. Restoration of the wild-type phenotype (e.g., reversal of a GFP expression shift) confirms the gene's function.
- Small Molecule or RNAi Inhibition: If possible, use pharmacological inhibitors or siRNA/shRNA to suppress the target gene's activity and assess if the phenotype is reproduced.
From Validation to Mechanistic Insight
After a hit is validated, the focus shifts to understanding its mechanistic role. Integrating single-cell RNA sequencing with CRISPR screening (Perturb-seq) allows for the transcriptomic characterization of cells bearing specific sgRNAs [12] [98]. In a GFP-based screen, one can sort cells based on GFP intensity (high vs. low) and subject them to scRNA-seq. This reveals how the genetic perturbation alters global gene expression patterns, potentially illuminating the signaling pathways and biological processes through which the candidate gene operates.
Diagram: Integrating scRNA-seq for mechanistic insight.
The Scientist's Toolkit: Research Reagent Solutions
The table below lists essential materials and reagents required for executing the CRISPR screening and validation workflows described in this note.
Table 2: Essential Research Reagents for CRISPR Screening & Validation
| Reagent / Tool | Function | Application Notes |
|---|---|---|
| CRISPR Library [97] | Delivers a pool of sgRNAs for high-throughput screening | Can be genome-wide or focused; available as lentiviral-ready plasmid pools or pre-packaged lentivirus. |
| Cas9-Expressing Cell Line | Provides the nuclease for CRISPRko screens | Can be generated by stable transduction. For CRISPRi/a, a cell line expressing dCas9-fusion proteins is required [12]. |
| Lentiviral Transfer Plasmid | Vector for cloning and delivering individual sgRNAs | Should contain a selection marker (e.g., puromycin) or reporter (e.g., GFP). |
| Non-Targeting Control sgRNAs [97] | Critical negative controls | sgRNAs with no known target in the genome, used to establish baseline phenotype and for normalization. |
| Flow Cytometry Panel [100] | Analyzes and sorts cells based on markers like GFP | Multiparameter panels require careful antibody titration and compensation controls to accurately resolve GFP-positive populations. |
| Viability Dye [100] | Discriminates live/dead cells | Essential for excluding dead cells that can non-specifically bind antibodies and confound analysis. |
The path from a primary CRISPR screen to a validated, biologically relevant hit is intricate. By employing a stratified strategy that combines rigorous bioinformatics, cross-referencing with public data, and, most importantly, experimental validation using individual sgRNAs and orthogonal CRISPR modalities, researchers can dramatically increase their confidence in screening results. Integrating these validated hits with advanced functional genomics tools like single-cell transcriptomics ultimately paves the way for a deeper mechanistic understanding of gene function in health and disease.
Conclusion
GFP-based visual screening represents a powerful and accessible method for identifying CRISPR transformants, particularly valuable in high-throughput applications and functional genomics studies. When implemented with careful attention to system design, optimization, and multi-layered validation, this approach can significantly accelerate the pace of gene function discovery and therapeutic target identification. The integration of GFP screening with emerging technologies—including improved Cas enzymes with higher fidelity, advanced delivery systems like lipid nanoparticles, and sophisticated bioinformatics pipelines—promises to further enhance its utility. As CRISPR applications expand into more complex disease modeling and clinical development, robust visual screening methodologies will remain essential for validating editing efficiency and understanding phenotypic consequences. Researchers should view GFP screening not as a standalone technique but as a component of a comprehensive validation strategy that leverages multiple orthogonal methods to ensure reliable, reproducible results in both basic research and therapeutic development.
References
- 1. Green fluorescent protein is a quantitative reporter of gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1242169/]
- 2. A reporter system for enriching CRISPR/Cas9 knockout ... [https://www.nature.com/articles/s41598-021-91760-9]
- 3. Fluorescent CRISPR Reporters: SRIRACCHA and ... [https://blog.addgene.org/fluorescent-crispr-reporters-sriraccha-and-gemcherry2]
- 4. Rapid and Robust Generation of Homozygous Fluorescent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12346671/]
- 5. Expanding the application of a UV-visible reporter for ... [https://www.nature.com/articles/s41438-021-00663-3]
- 6. Development and Validation of an Effective CRISPR/Cas9 ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2018.01533/full]
- 7. Evidence for expression of promoterless GFP cassette [https://pmc.ncbi.nlm.nih.gov/articles/PMC6714119/]
- 8. A native visual screening reporter-assisted CRISPR/Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12131447/]
- 9. Identification of Glyceraldehyde-3-Phosphate Dehydrogenase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6770653/]
- 10. Elevated expression of exogenous RAD51 enhances the ... [https://www.bmbreports.org/journal/view.html?uid=1774&vmd=Full]
- 11. A systematic comparison of CRISPR-Cas9 allele editing in ... [https://www.nature.com/articles/s41598-025-19115-2]
- 12. Large-scale CRISPR screening in primary human 3D ... [https://www.nature.com/articles/s41467-025-62818-3]
- 13. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 14. Protocol to rapidly screen CRISPR-Cas9 gene editing ... [https://pubmed.ncbi.nlm.nih.gov/40674218/]
- 15. Optimized delivery of dual-gRNA CRISPR/Cas9 via ... [https://www.sciencedirect.com/science/article/abs/pii/S2452014425002237]
- 16. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 17. Development of an RNA aptamer-assisted CRISPR/Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12614593/]
- 18. Fluorescent Bacterial Colony Selection using QPix 400 [https://www.moleculardevices.com/en/assets/app-note/bpd/fluorescent-bacterial-colony-selection-using-qpix-400-system]
- 19. FAST, a method based on split-GFP for the detection in ... [https://www.nature.com/articles/s41598-024-58588-5]
- 20. Quantitative measurement of cell-surface displayed proteins ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02386-1]
- 21. Green/red fluorescent protein disrupting drugs for real‐time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12079514/]
- 22. CRISPR: A Screener's Guide - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7036479/]
- 23. Fluorogenic CRISPR for genomic DNA imaging [https://www.nature.com/articles/s41467-024-45163-9]
- 24. 14 Comparing quantitative data between individuals [https://bookdown.org/pkaldunn/SRM-Textbook/BetweenQuantData.html]
- 25. Fluorescence activated cell sorting (FACS) in genome-wide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6386612/]
- 26. Simple Autofluorescence-Restrictive Sorting of eGFP+ RPE ... [https://www.frontiersin.org/journals/drug-delivery/articles/10.3389/fddev.2022.898568/full]
- 27. FACS-Based Functional Protein Screening via Microfluidic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7311539/]
- 28. Considerations for high-yield, high-throughput cell ... [https://www.nature.com/articles/s41598-018-36698-1]
- 29. Optimization of pre-enrichment strategies for mouse ... [https://www.sciencedirect.com/science/article/pii/S0301472X24004478]
- 30. Factors Influencing Fluorescence-activated Cell Sorting for ... [https://pubmed.ncbi.nlm.nih.gov/40130307/]
- 31. CRISPR libraries: A powerful screening tool for function ... [https://www.sciencedirect.com/science/article/pii/S3050538025000389]
- 32. Protocol to rapidly screen CRISPR-Cas9 gene editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12282252/]
- 33. A benchmark comparison of CRISPRn guide-RNA design ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11386-3]
- 34. A roadmap toward genome-wide CRISPR screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11960495/]
- 35. Pooled multicolour tagging for visualizing subcellular ... [https://www.nature.com/articles/s41556-024-01407-w]
- 36. CRISPR Sequencing [https://dnacore.mgh.harvard.edu/new-cgi-bin/site/pages/crispr_sequencing_main.jsp]
- 37. Protocol for generation of CRISPR-Cas9-mediated specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11388652/]
- 38. Perturb-Multimodal: A platform for pooled genetic screens ... [https://www.sciencedirect.com/science/article/pii/S0092867425005720]
- 39. Applications of multiplexed CRISPR–Cas for genome ... [https://www.nature.com/articles/s12276-025-01500-6]
- 40. GFP-on mouse model for interrogation of in vivo gene editing [https://www.nature.com/articles/s41467-025-61449-y]
- 41. CRISPR-Cas9-based one-step multiplexed genome ... [https://pubmed.ncbi.nlm.nih.gov/39995681/]
- 42. Application of a multiplex CRISPR/Cas9 strategy for ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1633104/full]
- 43. Quantitative and automated high-throughput genome-wide ... [https://pubmed.ncbi.nlm.nih.gov/22395785/]
- 44. Genome-wide CRISPR screen identifies Menin and SUZ12 ... [https://www.nature.com/articles/s41556-025-01751-5]
- 45. A MODULAR PLATFORM FOR DIFFERENTIATION OF ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5737635/]
- 46. Advances in human pluripotent stem cell reporter systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11407076/]
- 47. Absolute protein quantification using fluorescence ... [https://www.nature.com/articles/s41467-022-34232-6]
- 48. Rapid Generation of Marker -Free P. falciparum... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0168362]
- 49. a web-based interactive tool to visualize CRISPR screening ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04275-5]
- 50. Unlocking the potential of CRISPR tools and databases for ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1563711/full]
- 51. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 52. Ensure Proper Controls in Your CRISPR Experiments [https://www.synthego.com/blog/controls-crispr-experiments/]
- 53. a visualization toolbox to enhance selection efficiency in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8131052/]
- 54. Optimizing viral transduction in immune cell therapy ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06524-0]
- 55. A review of emerging physical transfection methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7196308/]
- 56. Viral and non-viral vectors in gene therapy: current state and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12271757/]
- 57. Improving viral-based gene delivery to mammalian cells ... [https://www.sciencedirect.com/science/article/abs/pii/S0022354925003727]
- 58. Non Viral Transfection Reagents Market Size, Share 2025- ... [https://www.coherentmi.com/industry-reports/non-viral-transfection-reagents-market]
- 59. Progress and challenges in viral vector manufacturing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4802372/]
- 60. Enhancing non-viral gene delivery to human T cells ... [https://www.sciencedirect.com/science/article/pii/S0753332225000149]
- 61. Rapid Assessment of CRISPR Transfection Efficiency and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8978543/]
- 62. CRISPR Controls for Genome Editing [https://www.thermofisher.com/us/en/home/life-science/genome-editing/crispr-controls.html]
- 63. DNA damage repair: historical perspectives, mechanistic ... [https://www.nature.com/articles/s41392-021-00648-7]
- 64. Comprehensive interrogation of synthetic lethality in the ... [https://www.nature.com/articles/s41586-025-08815-4]
- 65. Validation Study to Determine the Accuracy of Widespread ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9164083/]
- 66. Weaponizing CRISPR/Cas9 for selective elimination of ... [https://www.sciencedirect.com/science/article/pii/S1568786425000369]
- 67. WormSNAP: A software for fast, accurate, and unbiased ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599913/]
- 68. Optimizing the Protein Fluorescence Reporting System for ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.825212/full]
- 69. A high-efficient and naked-eye visible CRISPR/Cas9 ... [https://link.springer.com/article/10.1007/s00425-022-04060-5]
- 70. A model for accurate quantification of CRISPR effects in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11213010/]
- 71. Two-step CRISPR-Cas9 protocol for transposable element ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10440348/]
- 72. Troubleshooting Low Knockout Efficiency in CRISPR ... [https://www.biosynsis.com/blog/troubleshooting-low-knockout-efficiency-in-crispr-experiments/]
- 73. How to Pick the Best CRISPR Data Analysis Method for ... [https://www.synthego.com/guide/how-to-use-crispr/analysis-methods/]
- 74. Efficiency Optimization of CRISPR/Cas9-Mediated ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2019.00612/full]
- 75. Methods for Optimizing CRISPR-Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4976696/]
- 76. Validating CRISPR/Cas9-mediated Gene Editing [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/advanced-gene-editing/validating-crispr-cas9-mediated-gene-editing?srsltid=AfmBOoo1vNfTiBnk3YGAyN1O7Q3GnfGjOLHOr0Q-S7hP9GMY6x6vFPSc]
- 77. Validating CRISPR: How to Confirm Successful Editing [https://bitesizebio.com/47275/validate-crispr-experiment/]
- 78. Validation of CRISPR-based Gene Editing [https://www.neb.com/en-us/products/crispr-cas-gene-editing/crispr-validation?srsltid=AfmBOoo-djrURjEv_oegQoFWvqtdMqvpbWjCkTNGjqQ2z91cYTtLWn-u]
- 79. TALEN and CRISPR Validation Tools [https://www.thermofisher.com/us/en/home/life-science/genome-editing/crispr-validation.html]
- 80. Next Generation Sequencing Validating Your CRISPR ... [https://www.cd-genomics.com/next-generation-sequencing-validating-your-crispr-cas9-edit.html]
- 81. High-Throughput Genotyping | CRISPR Validation [https://www.genewiz.com/public/services/next-generation-sequencing/high-throughput-genotyping]
- 82. Quantification of GFP signals by fluorescent microscopy ... [https://pubmed.ncbi.nlm.nih.gov/24841297/]
- 83. Detection of GFP by flow cytometry using Anti-GFP antibodies [https://www.jacksonimmuno.com/technical/products/groups/anti-gfp/flow]
- 84. A novel GFP-based strategy to quantitate cellular spatial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11481894/]
- 85. Techniques for Validating CRISPR Changes Using RNA- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12027141/]
- 86. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 87. Revolutionizing Gene Therapy Protein Expression Potency ... [https://www.genengnews.com/sponsored/revolutionizing-gene-therapy-protein-expression-potency-assays/]
- 88. CRISPR Transfection Protocols Guide: How To Select The ... [https://www.synthego.com/guide/how-to-use-crispr/transfection-protocols/]
- 89. Establishing an auxin-inducible GFP nanobody-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641487/]
- 90. Comparative Analysis of Methods for Assessing On-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932317/]
- 91. A Highly Sensitive GFP Activation Assay for Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8636026/]
- 92. Rapid Assessment of CRISPR Transfection Efficiency and ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2022.854866/full]
- 93. Head-to-head Comparison of Green Fluorescent Protein ... [https://pubmed.ncbi.nlm.nih.gov/38925805/]
- 94. Adding a Chemical Biology Twist to CRISPR Screening - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10427134/]
- 95. Decoding CRISPR Screening: Methods, Applications and ... [https://www.cd-genomics.com/biomedical-ngs/resource/crispr-screening-workflow-advantages-application.html]
- 96. Research Techniques Made Simple: CRISPR Genetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8525197/]
- 97. Introduction to CRISPR screening | IDT [https://eu.idtdna.com/page/support-and-education/decoded-plus/overview-what-is-crispr-screening/]
- 98. Bioinformatics approaches to analyzing CRISPR screen data [https://pmc.ncbi.nlm.nih.gov/articles/PMC10019185/]
- 99. The Open Repository of CRISPR Screens [https://blog.addgene.org/the-open-repository-of-crispr-screens]
- 100. Best Practices for Multiparametric Flow Cytometry [https://www.thermofisher.com/us/en/home/references/newsletters-and-journals/bioprobes-journal-of-cell-biology-applications/bioprobes-79/best-practices-multiparametric-flow-cytometry.html]